Ashutosh Saxena1, Archita Patel2* and Priti Mehta1
1Institute of Pharmacy, Nirma University, Ahmadabad, Gujarat, India
2K. B. Institute of Pharmaceutical Education and Research, Gandhinagar, Gujarat, India
*Corresponding Author: Archita Patel, Assistant Professor, Department of Pharmaceutical Chemistry and Analysis, K. B. Institute of Pharmaceutical Education and Research, Gandhinagar, Gujarat, India.
Received: March 15, 2021; Published: April 05, 2021
The use of monoclonal antibodies is increasing in treatment of various autoimmune diseases, and hence knowledge about bioanalytical methods for the same is a need of the hour. Ligand binding assays (LBAs) have been traditionally used in the bioanalysis of mAbs due to its specificity and ease of sample preparation. Although they are still considered as a gold standard, technological advancement in LC-MS configuration has led to its widespread utilization as a bioanalytical tool. Further, LC-MS/MS has led to the development of more accurate and specific methods with respect to multi-component matrices. The present review encompasses discussion and comparative assessment of the method development strategies, validation parameters, merits and demerits of LBA and LC-MS techniques used for bioanalysis of mAbs. Recent advancements such as 2D-LC-MS/MS and future prospects of hybrid LBA-LC-MS approach are also included. The technologies and analytical strategies presented here will undoubtedly help readers in selection of methods for bioanalysis of novel biological entities (NBE) or biosimilars for preclinical and clinical studies in drug development.
Keywords: LBA; LC-MS/MS; Monoclonal Antibody; Bio-analytical Methods
Bioanalysis is a branch of analytical chemistry that deals with the quantitative estimation of xenobiotics (drugs and their metabolites) in biological fluids such as blood, serum, urine, etc. It involves a set of procedures which include collection, processing, storage and analysis of biological matrix for a selected compound. Bioanalysis is an integral part of the drug development process, as it is used for the purpose of performing pharmacokinetic, toxicokinetic, bio-equivalence and exposure responses like pharmacokinetic/pharmacodynamic studies. The determination of drugs in biological fluids is essential for therapeutic drug monitoring (TDM), forensic and clinical toxicology as well as pharmacology related research [1]. It helps to deal with issues such as exposure-response relationships, drug-drug interactions and can also be used as a tool to evaluate patient adherence to daily therapy [2]. It is also conducted to improve therapeutic efficacy and reduce any side effects of both small and large molecule drugs. Due to the wide-ranging dynamics of bio-analysis, the analytical techniques used for determination in routine analysis should be simple in performance, suitable for automation, accurate, rapid, selective and sensitive [1]. However, bio-analysis of large molecule drugs i.e. mAbs, is more challenging due to their high molecular weight, complex structure and a set of complicated interactions with other macromolecules of the body. Apart from that, development and validation of a bioanalytical method is also an important part for biosimilar product development (BPD), such bioanalytical methods should be capable of reliably and accurately measuring both proposed biosimilar and reference product in a biological matrix.
Before we talk about the analytical methods for bioanalysis of mAbs, it is necessary that we shed light on the structural parameters that play an important role in the same.
Monoclonal antibodiesMonoclonal antibodies (mAbs) are glycoproteins of high molecular weight which primarily resemble the gamma immunoglobulin (IgG). IgGs are further classified into 4 subgroups, namely IgG1, IgG2, IgG3 and IgG4 based on heavy chain sequences and interchange disulfide bond patterns. The groups of IgG1, IgG2 and IgG4 are widely used as mAbs, whereas IgG3 is rarely used due to their unusually short serum half-life.
mAbs are primarily used for targeted therapy and their success is mainly owed to their long serum half-life along with their target selectivity and specificity abilities. The diminished risk of unwanted immunogenicity due to their similarity in the sequences of chimeric or humanized mAbs with human mAbs makes them an ideal choice of therapeutic proteins [3].
The mAbs of therapeutic importance are with a high degree of heterogeneity including charge variants, aggregates, fragments and post-translational modifications (PTMs). Common PTMs like glycosylation, deamidation, oxidation, glycation, etc. can be introduced over the life span of mAbs during production, storage and in-vivo circulation by chemical or enzymatic modifications which have the potential to impact the safety and/or efficacy of mAbs [4].
Large molecular size, complex structure and complicated interactions with other molecules in the body provide mAbs with unique pharmacokinetics. The extent and rate of absorption of mAbs is variable and is affected by factors like size and charge of molecule, formulation and pre-systemic proteolytic degradation. Due to their prominent susceptibility to gut proteases, these drugs are administered intravenously, subcutaneously or intramuscularly. Endothelial cells or monocytes take up the circulating IgGs either by fluid-phase pinocytosis or receptor-mediated endocytosis following which, IgGs bind to FcRn in the endosome and transported back to the cell surface where they are then released under neutral pH conditions and this mechanism is primarily responsible for the long half-life exhibited by IgGs. When proteins are not bound to FcRn, they are sorted to the lysosomes for degradation. Deamination, oxidation, glutamate/pyroglutamate conversion and C-terminal lysine clipping are the biotransformation events that have been commonly observed with therapeutic antibodies. Glomerular filtration by kidneys is not the mechanism for elimination of these proteins owing to their large size. Endocytosis and proteolytic degradation collectively known as Target-Mediated Drug Disposition (TMDD) is the most significant pathway for elimination of these mAbs [5].
During bioanalysis of mAbs, analytical methodology should differentiate mAbs and endogenous IgG with respect to the complementary determining region (CDR). Also, the formation of antidrug antibodies (ADA) can interfere with accurate quantification of therapeutic proteins leading to compromised pharmacokinetic data interpretation [6].
The commonly used bioanalytical methods for mAbs can be classified into two categories: (1) Ligand binding assays (LBAs) and their various modifications and (2) Liquid chromatography-mass spectrometry (LC-MS) with different modes of detection.
We tried to compare working modalities, advantages and disadvantages of both techniques with respect to its use for bioanalysis of mAbs. Additionally, recent updates about hybrid techniques are also discussed here.
Ligand binding assays (LBAs) as a bioanalytical toolThe scientists Rosalyn Yalow and Solomon Berson reported the first successful ligand binding assay in 1960 for investigating insulin and insulin specific antibody interaction [7]. As the name suggests, ligand binding assays are based on the binding interactions with other biological molecules. It involves critical reagents most commonly used are monoclonal or polyclonal antibodies. They are considered as critical because the quality, selectivity, and stability of such reagents can affect the method performance or output of the method that binds to the analyte of interest. The preferred critical reagents most commonly used are monoclonal or polyclonal antibodies.
The most common LBAs used for bioanalysis of antibodies are Enzyme linked immunosorbent assay (ELISA). Engvall and Pertman were the first to describe an ELISA method in 1971 for the quantification of rabbit immunoglobulin G (IgG) [8].
The most commonly used ELISA format for bioanalysis of therapeutic mAbs is the sandwich ELISA format. In this format, firstly, a catcher specific for mAb of interest, referred to as primary catcher is coated on the walls of the titer plate. In the second step, a pre-diluted biological sample consisting of mAb is added to the plate and hence the analyte will bind to the primary catcher. Subsequently, the plates were washed and the therapeutic mAb bound to the primary catcher is identified by a secondary catcher specific for the same mAb which, in most cases, is an anti human IgG. To this secondary catcher, an enzyme is attached directly or indirectly with the help of a conjugate specific for the enzyme. Finally, a chromogenic substrate is added which is converted by the enzyme into a colored soluble product. The colored reaction can be stopped by the addition of a strong acid. The colored product is then analyzed by a spectrometric plate reader. The most commonly used enzymes are HorseRadish Peroxidase (HRP) or Alkaline Phosphatase (ALP). HRP acts by catalyzing the oxidation of substrates such as 3, 3’, 5, 5’-tetramethyl benzidine (gives blue colored product) or o- phenylenediamine dihydrochloride (gives yellow-orange product). ALP catalyzes the hydrolysis of phosphate groups from the substrate molecules like p- nitrophenyl phosphate which gives a yellow reaction product. Bioanalysis of mAbs can be done with other immunoassay formats such as immune-fluorescence assays using flow cytometry and radio-LBAs. Both these assays operate in a manner similar to ELISA that is, using a primary and secondary catcher. However, unlike enzymes that are used in ELISA, immune-fluoroscence assays used a fluorescent label, whereas, radio-LBAs used a radioactive label on secondary catcher for quantification of therapeutic mAbs [9].
The workflow for different LBA formats is shown in figure 1. The different LBA formats for the bioanalysis of mAbs (upto 2009) have been reported by Damen., et al. and the same have been represented in the form of a pie chart in figure 2 [9]. It can be concluded that different formats have been shown to be effective for the bioanalysis of a given mAb and the choice of a particular format depends on the sensitivity and ease of methodology, among other factors offered by each format.
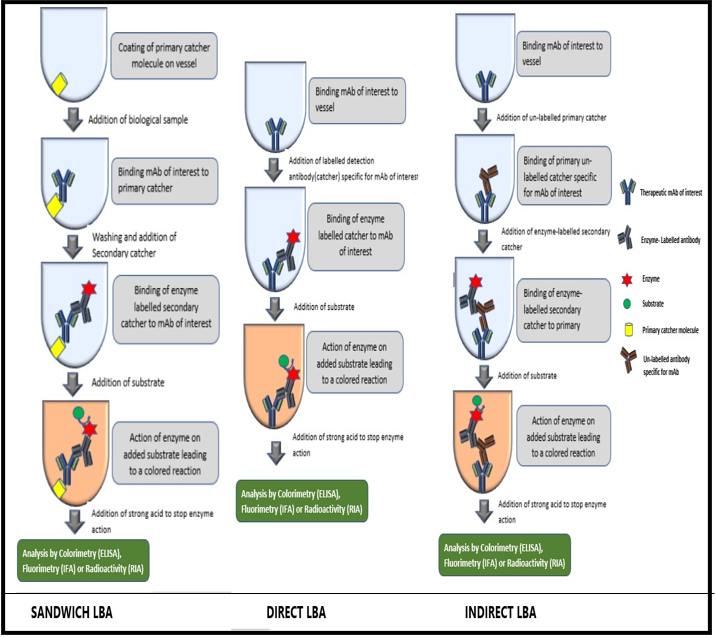
Figure 1: Workflow for bioanalysis of mAbs using different LBA formats.
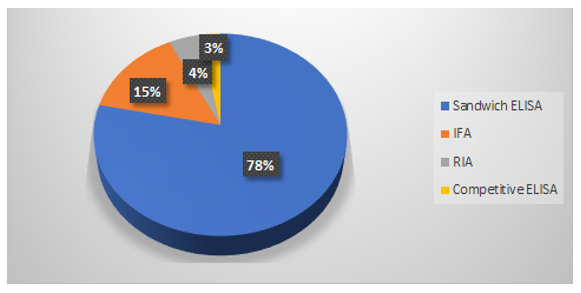
Figure 2: Percentage distribution of different LBA formats for bioanalysis of mAbs.
LBAs offer diverse applicability in bioanalysis due to benefits like low cost, high sensitivity and selectivity in bioanalysis. In spite of their widespread applicability, LBAs have prominent issues that need to be dealt with which include inhibition from competitive molecules and matrix and indirect detection of antibody molecules.
Matrix effects can be considered as an important factor to control during bioanalysis of mAbs. They can be classified as specific and non-specific matrix effects. Non-specific binding proteins like rheumatoid factor and heterophilic anti-IgG antibodies are a couple of factors that non-specifically interfere with antigen-antibody reactions of LBAs. On the contrary, proteases, anticoagulants and soluble drug targets are some factors that may cause specific interference. These matrix effects make it substantially difficult to maintain adequate assay sensitivity and reproducibility. This makes the development of a validated immunoassay method challenging to accomplish. Hence, these matrix effects need to be identified and mitigated through sample pretreatment steps, extraction of known interferences or through the addition of buffer reagents [10]. In addition, there are certain components that have the potential to interfere and pose a significant challenge on the utility of LBAs for bioanalysis. Anti Drug antibodies (ADA) are endogenous antibodies that are produced in response to unwanted immunogenicity reactions. These antibodies affect the accuracy of LBAs. In their presence, only the free form of the drug is measured since the therapeutic mAbs trapped by ADA are incapable of being quantified [11]. An example of this type of interference was reported by Preissner., et al. wherein thyroglobulin a thyroid biomarker led to the underestimation of thyroglobulin concentration [12]. Endogenous proteins such as insulin, growth hormone and recombinant growth factors may bind to circulating receptors with substantial variability in concentrations inter-individually. Due to this variability, the pharmacokinetic properties are modified thereby, acting as interferences in LBAs. Susceptibility of mABs to structural modulations may implicate loss of biological activity and varied pharmacokinetic properties [11]. One such example is the non-enzymatic deamidation of asparagine and glutamine residues during manufacture [13].
Also, analytically significant non-specific binding upon application of a sample to a plastic surface, such as polystyrene microtiter plates is another potential source of interference in LBAs. This phenomenon shows high inter-subject variability and is influenced by a variety of assay conditions such as type of microtiter plate, washing procedures, etc. [11].
In lieu of the shortcomings inherent in ligand binding assays (LBA), chromatographic techniques, especially LC-MS have been considered as an alternative and have been applied to the bioanalysis of mAbs in preclinical as well as clinical studies.
LC-MS as a bioanalytical toolIn lieu of the shortcomings inherent in ligand binding assays (LBA), chromatographic techniques, especially LC-MS have been considered as an alternative and have been applied to the bioanalysis of monoclonal antibodies in preclinical as well as clinical studies. Bioanalysis of mAbs using LC/MS is a newer approach compared to LBA.
Compilation of various published research articles through Pubmed, Google scholar, and Elsevier, the bio-analysis of monoclonal antibody using LC/MS since last two decades could be divided into two major parts: (1) Analysis of mAb after digestion and (2) Intact mAb analysis. The most common approach is quantitation of mAb through enzymatic digestion followed by analysis of one or multiple selected surrogate peptides using LC-MS spectrometer as shown in figure 3.
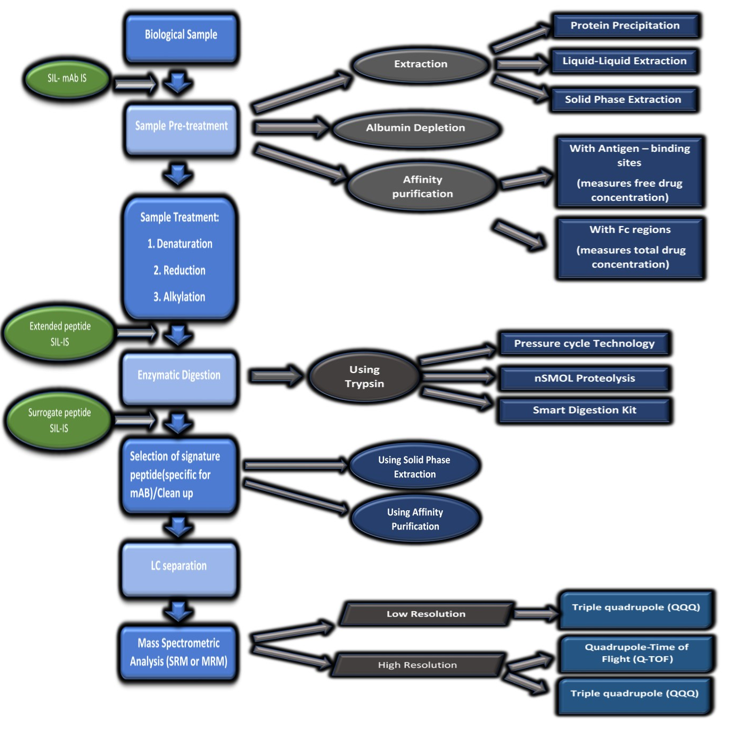
Figure 3: Workflow for bioanalysis of mAbs using LC-MS.
Quantification of therapeutic mAbs can be divided into three proteomics approaches, bottom‐up, middle down and top‐down approach (Figure 4). The bottom‐up approach involves digestion of protein into small peptides known as surrogate peptides and analyzing such peptides by LC-MS/MS using a triple quadrupole instrument hence this technique has a major advantage of higher sensitivity. Therefore bottom up approach is a widely used technique for bioanalysis of mAbs (R). Figure 5 shows the proportion of different LC-MS approaches used for bioanalysis of mAbs [14].
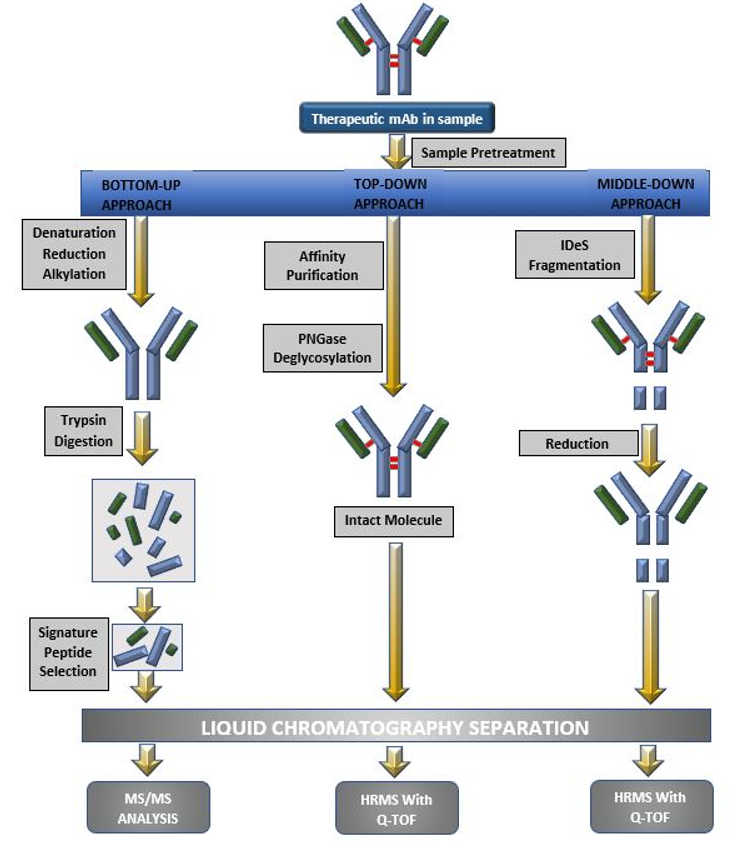
Figure 4: Different LC-MS approaches (a. Bottom-up approach; b. Top-down approach; c. Middle-up approach).
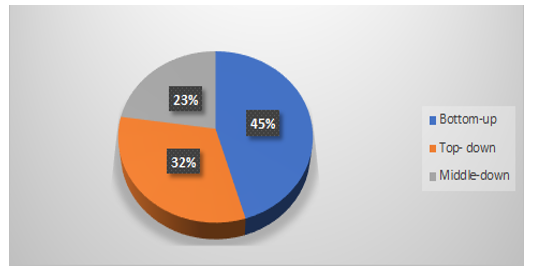
Figure 5: Percentage distribution of different LC-MS approaches for bioanalysis of mAbs.
The bottom up approach represents a primary question: whether selected surrogate peptide can represent the complete picture of chosen mAb? The major requirements for surrogate peptides are that they should be unique to the target protein analyte and should be free from interference at Selected Reaction Monitoring (SRM) transitions. Tryptic peptides of conservative sequence from constant regions of 1ight chain or heavy chain have been commonly used as surrogate peptides for monoclonal antibodies quantitation in case of prec1inical studies, whereas, in case of clinical studies, unique peptides from variable region, most usually the CRR are monitored which helps avoid interferences from endogenous human IgGs. The selection of surrogate peptides, peptide location on the protein and residence in the determining region is important [6]. The use of bottom-up approach allows collection of information about only a certain part of protein structure, as the selected surrogate peptide cannot give an idea about complete protein structure.
The top-down approach also known as the intact mass measurement method, employs high end quadrupole time of flight (Q-TOF) mass spectrometers and advanced mass deconvolution algorithms in order to measure the molecular weights of intact monoclonal antibodies with high accuracy. The method is simple to execute and also does not involve steps for selection of surrogate/representative peptide. It has been used in the bioanalysis of trastuzumab, bevacizumab with infliximab, Trastuzumab with MK-8226 and Anti-MUC16 mAb- monomethyl auristatin E (digestion with PNGase F) and bevacizumab [15-18].
In order to minimize the variability in chromatography and MS-ionization, a synthetic stable isotope labelled internal standard (SIL-IS) is required. Use of SIL-IS balances the variations in assay due to immunoaffinity capture, washing and elution steps, sample‐to‐sample matrix difference and instrument variations. Different SIL-IS used in development of bioanalytical methods of mAbs are given in table 1. Though, the use of the whole monoclonal antibody uniformly labelled with a stable isotope amino acid would be an ideal IS, the time and resources required to generate such an SIL-IS for each new candidate is huge and therefore avoided [19].
| Internal standard | Features | Advantages | Disadvantages |
|---|---|---|---|
SIL-Protein |
Prepared by recombinant protein synthesis and includes digestion of SIL-peptides |
Chemically synthesized and have higher resemblance to analyte and provides control throughout processing and analysis |
Requires biological synthesis for structural confirmation, time consuming, may have subtle differences than analyte (PTMs) |
SIL-Peptide |
Added after or before digestion |
No specific advantages |
Not appropriate with more selective extractions Corrections for recovery and/or ion suppression effects may be limited |
Extended SIL-peptide |
Extra flanking of aminoacids (typically 3-6 non-labeled) added to N- and/or C- terminus of SIL-peptide during synthesis, required to be added before digestion, “wings” are cleaved, leaving SIL-peptide |
Chemically synthesized and LC-MS/MS analysis can be performed under controlled conditions |
Can’t control protein analyte extraction/enrichment or digestion efficiency |
Non-SIL |
Structural analogue, added after digestion, needs to be similar enough with close chromatographic elution to target peptide (S) |
May provide some control for LC-MS/MS steps |
Can’t control protein analyte extraction/enrichment or digestion efficiency And limited Compensation for ion suppression effects |
Table 1: Different internal standards (IS) used in LC-MS bioanalysis.
The top-down approach has limited sensitivity as multiple peaks appear in both the chromatogram and the MS spectrum depending on the diversification of sugar chain structure. The utility of the middle-down proteomics approach of converting therapeutic mAbs into high-homogeneity and lower molecular weight fragments such as Fab, Fc, Lc, Fc/2, Fd by enzymatic cleavage and/or disulfide bond reduction before LC-MS analysis and quantifying them using high-resolution mass spectrometry(LC-HRMS) has been reported. The mAbs adalimumab, rituximab, trastuzumab MK-822 and eculizumab have been analyzed using middle down approach.
Apart from that it is also important to know about different LC-MS configurations used in bioanalysis of mAbs. Brief of each is discussed below.
After sample pretreatment and digestion, the processed samples are subjected to separation on a HPLC system. Reports suggest that C8 or C18 chromatographic columns are used in the majority of the procedures along with gradient elution to ensure effective separation [9].
LC-MS -SRM formats that incorporate CDR-surrogate peptides prove to be a valuable bioanalytical tool for quantitative analysis in non-clinical as well as clinical studies [20]. The LC-MS - SRM format by universal surrogate peptide approach uses a common tryptic peptide belonging to the CDR region. The dual universal peptide approach uses 2 surrogates from heavy and light chains. This provides us with enhanced information about the structural integrity of the analyte. MS detection using SRM mode offers us with higher sensitivity, better quantitative accuracy and a wide dynamic range for quantification of mAbs. It also allows for easy switching among different precursor/product ion transitions.
LC-HRMS is another MS detection technique that couples high resolution mass spectrometry with liquid chromatography. In order to conduct analysis by HRMS, one of the two methodologies have to be followed: a full scan accurate acquisition of all parameters reaching the mass analyzer without the application of any prior mass filter, or, a full scan accurate mass acquisition of the MS/MS product ions of certain selective parent precursor ions. Monoclonal antibodies exhibit multiple charged states and the response ratios of these charged states are inversely proportional to analyte concentrations. With the use of LC-HRMS, the ions exhibiting multiple charged states can be monitored and added up which in turn, widens the linear dynamic range. Additionally, HRMS provides the ability to select an isotope ion thereby, providing better selectivity and sensitivity. The advantage of post-acquisition data mining is another aspect that makes LC-HRMS suitable for bioanalysis. Thus, LC-HRMS is capable of not only identifying the analyte of interest, but also other compounds that may be important. An added advantage of HRMS is that it does not require any additional effort in relation to sample preparation, chromatographic optimization or sample injection [21].
Two dimensional LC-MS/MS (2DLC-MS/MS)Over the past 20 years there has been a revolutionary change in trends in bioanalysis of biotherapeutics, suggesting use of multidimensional LC to achieve greater resolution. The multiple reaction monitoring (MRM) assays are designed for measuring monoclonal antibody/protein in complex serum digests produced without analyte fractionation can be improved by multi-dimensional chromatography.
In 2D-LC, a usual separation is carried out in the first dimension and aliquots of the effluent are collected and injected to a second-dimension column that has very different separation selectivity compared to the first-dimension column like SCX × RPLC, HILIC × RPLC and RPLC × RPLC. SO, this technique utilizes two different separation principles in single run and hence much higher resolving power for separation of complex mixture and overall reduction in time required to fully separate simpler mixtures.
This technique can be performed online or off-line mode. The On-line 2D-LC can be of two types. In comprehensive 2D-LC (LC × LC) the whole stream of effluent of the first (1D) column is transferred to the second (2D) column. In heart-cutting LC (LC-LC), a peak or a part of the chromatogram is transferred to the second column. Heart-cutting LC in combination with MS is of particular interest in bio-analysis for the high sensitive measurement of target compounds in complex matrices. Multiple peaks or multiple parts of the chromatogram can also be selected for transfer to the second column [22,23].
Authors have recently described the quantitation of an IgE binding nanobody in cynomolgus monkey plasma by off-line 2D-LC-MS/MS. The method involved combined use of reversed-phase LC (RPLC) enrichment of a surrogate peptide (and internal standard peptide) at pH 10 (dimension 1) followed by RPLC- MS/MS (dimension 2). The developed 2D-LC-MS/MS was also well correlated to a ligand binding assay and similar pharmacokinetic parameters were estimated. Using the 2D-LC-MS/MS methodology, the limit of detection was 10 ng/mL. The value was indicative of at least two orders of magnitude improvement over direct LC-MS/MS analysis under identical LC-MS/MS conditions [24]. The other examples of mAbs that have been analyzed by 2D-LC-MS/MS includes IgG1 mAb designated ABC123 using triple quadrupole mass spectrometer [25] and myostatin using Q-Trap mass spectrometer in positive multiple reaction monitoring mode [26].
However, LC-MS methods pose several challenges as well. Proteins and peptides often generate multiple charged ions and exhibit complex mass spectra which results in sensitivity issues. Also, the lack of understanding of the nature of biotransformation and the impact it has on the quantitation data poses a significant challenge. Intrinsic challenges such as non-specific adsorption, matrix suppression, stability issues are very prominent. LC-MS methods are currently less sensitive than LBA assays which limit their use when the dose or exposure is low. In order to enhance sensitivity, extensive and highly effective sample preparation steps such as surfactant-aided precipitation, etc. must be applied to eliminate or mitigate matrix interferences. The interaction of additional variability due to the involvement of multiple complicated steps is another limitation of LCMS for bioanalysis. In order to improve the robustness and throughput of LC-MS assays, novel approaches such as microwave-assisted digestions, highly-efficient trypsin, etc. are being employed [19].
Bioanalytical method validation using LBA and LC-MSBioanalytical method validation is essential to ensure the acceptability of assay performance and the reliability of analytical results. Once the method has been developed, bioanalytical method validation proves that the optimised method is suited to the analysis of the study samples. Bioanalytical method validation improves the quality and consistency of the bioanalytical data in support of the development and market approval of Biotherapeutics. Recently ICH has published draft guideline M10 regarding bioanalytical method validation. Scope of guideline covers validation aspects of both types of analytical methods used for mAbs; LBA and LC-MS. A brief description of the assay validation parameters for LBA and LC-MS have been specified in table 2 [8].
| Element | LBA | LC-MS |
|---|---|---|
Basis of measurement |
Biological binding reaction |
Chemical properties of analyte |
Target concentration |
Free/bound/total concentration |
Total (free + bound) concentration only |
Sample preparation |
Not required |
Extensive |
Concentration response |
Non-linear |
Linear |
Detection |
Indirect |
Direct |
Calibration curve range |
Narrow range with <2 orders of magnitude |
Broad with several orders of magnitude |
Precision |
High |
Medium |
Specificity |
Comparatively less specific for discriminating drug from its metabolites |
Highly specific at molecular level and have the capacity for multiple component analysis |
Sensitivity |
Higher sensitivity (ng/mL) |
Comparatively less sensitive (µg/mL) |
Non-recurring costs |
Comparatively low |
High |
Recurring costs |
Comparatively high |
Low |
Internal standard |
Not required |
Required |
Table 2: Comparative assessment of LBA and LC-MS as bio-analytical tools.
Biological drugs like mAbs are highly complex analytes owing to their micro-heterogeneity. Complexity of bio-therapeutics is due to the production process which introduces multiple PTMs which lead to numerous isoforms. The generated isoforms differ with respect to their potency, clearance rates and other biological parameters [27]. Therefore, in order to develop an apt bioanalytical strategy, differentiation between the target binding components/active isoforms and inactive isoforms due to PTMs is highly essential. The intent of measuring the free form, bound form or both should be carefully known and this helps in generating a correct bioanalytical method. Few review papers and articles are available regarding use of both the techniques individually for bioanalysis of mAbs [8,9,27,28]. However, none of them has compared both the methods in context with its stepwise methodology development and validation aspects.
In case of LBA, it is assumed that capture antigen or anti-idiotype antibodies target the free drug whereas, the critical reagents target the Fc moiety of monoclonal antibody to measure the total drug. In capture assays (which involve specific entrapment), the therapeutic antibodies present in bound form in the sample may dissociate during incubation due to presence of large excess of capture reagent. Hence, it is highly likely that LBAs measure a mixture of bound and free forms which is highly dependent on assay parameters like concentration of coating ligand, incubation time and relative affinities. Apart from this, it is also possible that the induced endogenous antibodies may compete with anti-species antibodies used in the detection step in which case, neither total nor the free form is measured.
The LC-MS requires additional sample preparation steps such as extraction, affinity purification, and enzymatic digestion prior to bioanalysis of mAbs. The absence of such sample preparation steps in LBAs offers a fairly easy approach compared to LC-MS.
With the use of LC-MS technique, bioanalysis of mAbs, using top down approach, measures total drug while the immune-capture assay measures a mixture of free and bound forms same like classic LBAs. However, LC-MS based methods are capable of overcoming anti-antibody interferences by using either an appropriate labelled internal standard and/or by applying an enzymatic digestion step [28].
LBAs require critical reagents for each product and they also lack precision as compared to LC-MS/MS. Although LC-MS assays are able to achieve precise and reproducible results, internal standards are essential to get unbiased results [6].
LBAs use titer value (relative concentration) for the quantification of mAbs as its response is a nonlinear function of the analyte concentration. On the contrary, LC/MS is capable of giving absolute quantification of mAbs.
LC-MS pose significant challenges during bioanalysis of mAbs in terms of extensive, tedious sample preparation and pre-requisite expertise required in order to handle complex LC-MS instrumentation (Figure 3). On the other hand, LBAs have shown limited dynamics as they are subjected to antibody cross-reactivity and limited selectivity.
The difficulty in the determination of active drug exposure due to absence of a suitable pharmacodynamic or safety biomarker during total drug quantification is a limitation to LC-MS signature peptides approach as well as for generic LBA methods [27].
LC-MS assays can accommodate the industry requirements of high sensitivity, reproducibility and selectivity with the help of either Multiple Reaction Monitoring (MRM) or High Resolution (HR) quantification. Additionally, they provide the ability to measure multiple components [29]. A comparative assessment of the LBA and LC-MS [6] as a bioanalytical tool has been given in table 3.
| Parameters | LBA | LC-MS/MS (using a surrogate peptide, recommended) |
|---|---|---|
Calibration curve |
±20% of NC (non-zero calibrators) ±25% of NC (LLOQ and ULOQ); Non-linear curve; another points can be used |
±15% of NC (non-zero calibrators) ±20% of NC (LLOQ and ULOQ); Linear/Non-linear curve (depends on the assay method) |
LLOQ (%CV) |
± 25% |
± 25% |
Accuracy and Precision (RE,CV) |
Within 20% (LLOQ/ULOQ QCs within 25%).Minimum 6 runs |
Within 20% (LLOQ/ULOQ QCs within 25%).Minimum 3 runs |
Dilution integrity/Linearity |
CV within 20% |
CV within 20% |
Parallelism |
Dilution series CV within 30% incurred samples |
NA |
Selectivity/specificity |
10 Lots; LLOQ; accuracy within 25% for 80% of fortified lots |
6-10 Lots; blanks<20% of LLOQ or 5% of IS. LLOQ; accuracy within 25% for 80% of fortified lots |
Recovery |
NA |
Extracted vs post extracted samples of LQC, MQC and HQC. Results should be reproducible and consistent. |
Matrix stability |
Within 20% of nominal determined at each storage temperature |
Within 20% of nominal determined at each storage temperature |
Stock and working solution stability |
May not be required if covered by COA |
Old vs freshly prepared solutions; Mean values within 10% recommended |
Carryover(Blank following a ULOQ sample) |
NA |
Impact assessment needed; <20% of LLOQ |
NC- Nominal concentration; CV- Coefficient of variance; LLOQ- Lower limit of quantification; ULOQ-Upper limit of quantification; LQC- Lower quality control samples, HQC- High quality control samples, MQC- Middle quality control samples; COA- Certificate of analysis. |
||
Table 3: Assay validation parameters for LBA and LC-MS.
Although LBAs and LC-MS assays have significant aspects that qualify them as suitable tools for bioanalysis of mAbs, weakness of both techniques can be overcome by using a synergistic approach. This means, hybrid approach where in, the strengths of both the techniques can complement each other and provide better results. Hybrid approaches involve use of immune-affinity extraction procedures for selectivity and MS detection for sensitivity [27].
LBA/LC-MS hybridizationA combination of the enriched, purified sample by LBA and quantitative detection at the molecular level by LC-MS has given rise to a new platform, the hybrid LBA/LC-MS assay. The purification of the analyte of interest can be achieved by affinity-based methods mainly, immune-depletion and immune-capture. Immuno-depletion removes high-abundance serum proteins such as albumin, immunoglobulins, and transferrin whereas immune-capture isolates the analyte of interest from the biological matrix based on affinity. Isolated analytes further quantified using LC-MS platform. However, hybrid LBA-LC-MS assays show enhanced specificity, reduced drug interference and also encounter fewer false positive results [6]. Schematic diagram related to the workflow of Hybrid technique is illustrated in figure 6.
History of Hybrid assay developmentIn the early 1990s, the evaluation of an affinity capture technique was initiated by the Hutchens and Yip. There motive was to purify macromolecules by MALDI-TOF and referred to the process as “Surface enhanced affinity capture mass spectrometry”. Following this, Nelson., et al. worked on similar grounds using the term “Mass Spectrometry Immunoassay”. These steps proved to be ground-breaking in establishing and recognizing the synergism of both LBA and LC-MS for enhanced bioanalysis [30].
Hutchens and Yip were the first to evaluate affinity capture techniques for purifying macromolecules followed by quantification by MALDI-TOF Mass spectrometry in the early 1990s, referred to as “surface-enhanced affinity capture mass spectrometry” [7], followed by the work of Nelson et.al using the term “Mass Spectrometric Immunoassay” [8]. This work helped to prove the applicability of the Antigen-Antibody immuno-capture concept for identification and quantification of target antigens using mass spectrometry [30].
In 2014, Keyang Xu., et al. reported a hybrid LBA-LC-MS assay for the clinical pharmacokinetic assessment of 2 co-administered mABs, namely, mAb-A and mAb-B in human serum. The method omitted the need for any stringent affinity capture reagents and employed CDR peptides for each mAb as surrogate form Ab quantification. They concluded that not only are hybrid assays highly selective, they can be readily multiplexed as well [31].
However, few reports are available till date for the use of hybrid technology for bioassay of mAbs.Keyang Xu., et al. (2014) have reported a hybrid LC-MS/MS assay for simultaneous estimation of mAb-A and B in human serum. The mAbs were quantified using a signature peptide derived from each mAb as a surrogate. The method was able to efficiently develop a powerful quantitative tool for pharmacokinetic assessment of mAbs without the requirement for stringent affinity capture reagents [31].
In a report published by Kellie., et al. a basic hybrid LBA-LC-MS method has been described. However, instead of utilizing surrogate peptide approach for MS analysis, whole-molecule or larger fragment analysis was adopted. This enables the hybrid assay to achieve metabolism assessment in early preclinical studies or degradation in clinical studies [32].
In 2016, Authors have reported a whole-molecule immunocapture LC-MS quantitation of biotherapeutics that can be applied to preclinical/clinical in vivo samples using an anti-idiotypic antibody capture of a biotherapeutic monoclonal antibody (mAb) followed by its digestion with IdeS and disulfide reduction to produce fragments which were the analyzed directly by LC-MS [32].
In another attempt, Keyang Xu., et al. (2016), developed a validated immunoaffinity LC-MS method for mAb quantification in cynomolgus monkey serum. The method basically comprises of immunoaffinity capture of antibody in serum, enzymatic digestion and subsequent LC-MS detection of framework peptides. Additionally, similar assay conditions were used for six different mAbs in order to demonstrate the plug and play nature of hybrid assays [33].
Authors have developed the validation of an immunoaffinity (IA)-LC-MS/MS method for quantification of a Human/humanized mAb. IgG bio therapeutic molecules mAb-1 to mAb-7 (having different origin monkey, rat, mouse and human) bio-therapeutic in cynomolgus monkey serum under identical conditions. This method includes immune-affinity capture of the antibody in serum, followed by enzymatic digestion and detection of a framework peptide. This method was validated and found to have ‘plug and play’ nature [33].
The use of magnetic beads for immunocapture is one of the most widely used techniques in hybrid assays. However, magnetic beads are not capable of complete automation, prone to non-specific binding and are not cost-efficient. Chen., et al., attempted to determine an alternate and more suitable immunocapture approach in order to maximize sensitivity of hybrid assays. They used seven different ELISA plates and compared the results to a magnetic bed immunocapture LC-MS assay and a colorimetric ELISA [34].
In another report recently, in 2018 scientists have developed immunocapture-LC-MS hybrid assay for quantitation of BI123ABC from cynomolgus monkey plasma. Amongst seven different ELISA plates tested, streptavidin plates were found to be noteworthy. The hybrid assay performance was evaluated and compared with a magnetic bead-based immunocapture-LC-MS assay and a colorimetric ELISA. The hybrid assay was similar to the magnetic bead immunocapture-LC-MS assay and wider than the ELISA. ELISA plate immunocapture provided a cleaner sample than the bead immunocapture, resulting in a lower LLOQ [34].
Hybrid LBA-LC/MS assay platform provides the highest possible selectivity, sensitivity, and linear dynamic ranges, thus significantly improving detection of antibodies in bioanalysis [27]. However, cost, sample volume requirements, assay throughput, and difficulty in implementation continue to be limiting factors.
The paradigm shift from the use of small molecules to large molecules as therapeutics is happening exponentially. This makes it only necessary for the techniques required for characterization and quantification of large molecules in biological matrices to keep in pace with the advancement. Amongst the biologics used in the treatment of diseases; mAb being the major player demands the use of more dynamic techniques such as LC-MS. Although LBAs were considered as a gold standard for the bioanalysis of mAbs owing to their high sensitivity, LC-MS methodologies have been given considerable thought in recent years. LC-MS based bioassays offer dynamic applications in the field of discovery research, tissue sample analysis, and pharmacokinetic assessment. The decisive factors for the choice of a particular bioanalytical strategy may be availability of reagents, internal standards, and instrumental prowess among others. It is therefore essential both the techniques should go hand in hand in order to get a complete picture about the pharmacokinetic profile of mAbs. To overcome the loop holes of both the techniques, recently scientists have tried hybridization technique that involves immunocapture of desired mAbs with LBA followed by their chromatographic analysis using LC-MS analysis allowing better and more desirable results. It can be considered as a major revolution in the field of bioanalysis.
Citation: Priti Mehta., et al. “Comparison and Recent Updates of LBA and LC-MS/MS Methods for Bio-analysis of Therapeutic Monoclonal Antibodies". Acta Scientific Pharmaceutical Sciences 5.5 (2021): 13-25.
Copyright: © 2021 Priti Mehta., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
















 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
