Hande Z Tıras1, Faruk Incecik1*, Atıl Bisgin2 and Ozlem M Herguner1
1Department of Pediatric Neurology, Balcalı Hospital and Clinics, Deptartment of Medical Genetics and Cukurova University AGENTEM (Adana Genetic Diseases Diagnosis and Treatment Center, Adana, Turkey
2Cukurova University Faculty of Medicine, Balcalı Hospital and Clinics, Deptartment of Medical Genetics and Cukurova University AGENTEM (Adana Genetic Diseases Diagnosis and Treatment Center, Adana, Turkey
*Corresponding Author: Faruk Incecik, Department of Pediatric Neurology, Balcalı Hospital and Clinics, Deptartment of Medical Genetics and Cukurova University AGENTEM (Adana Genetic Diseases Diagnosis and Treatment Center, Adana, Turkey.
Received: February 13, 2024; Published: February 21, 2024
Citation: Faruk Incecik., et al. “Clinical and Genetic Evaluation of Epileptic Encephalopathy Cases in Turkey: A Single Centre Experience”. Acta Scientific Paediatrics 7.3 (2024): 17-24.
Objective: Early-onset epileptic encephalopathy (EOEE) is a grouping comprising many conditions that may severely disable children with developmental delay and severe epilepsy. They have variable genetic heterogeneity. In recent years, genetic causes have started to be detected more frequently in the ethology of EOEE. We aimed to determine the genetic and phenotypic relationship in eoee patients with the next-generation
Methods: Genetic studies were performed with a next-generation sequencing panel from 37 patients diagnosed with EOEE. For the study, the causal variants (pathogenic, likely pathogenic and VUS) detected in the patients were examined. The phenotype-genotype relationship of the patients was evaluated.
Results: No mutation could be detected in 10 (27%) of 37 patients. Pathogenic variant was detected in 5 (13.5%) patients, likely pathogen in 5 (13.5%) and VUS in 7 (19%) patients. Changes of unknown clinical significance were detected in 10 patients. In total, the causative variant (pathogenic, likely pathogenic and VUS) was detected in 17 (45.9%) patients. In this study, the diagnostic value determined by known gene panel analysis was 27% (P, OP) and 45.9% (P, OP, and VUS).
Conclusions: The diagnostic value found in our study was 27% (P, OP) and 45.9% (P, OP and VUS), which was consistent with recent studies using targeted gene panel analyzes in the diagnosis of patients with early-onset epileptic encephalopathy. The targeted gene panel is seen as a practical diagnostic tool for patients with EOEE, as it enables to establish genotype-phenotype correlations and guides treatment.
Keywords: Epileptic Encephalopathy; Genetic; Next Generation Sequencing
Early-onset epileptic encephalopathies (EOEE) are disorders that are caused by recurrent clinical seizures or prominent interictal epileptiform discharges and usually seen during the early infantile period. Approximately 40% of seizures occur during the first 3 years of life due to epileptic encephalopathies [1].The most common EOEE are Ohtahara syndrome (OS), epilepsy of infancy with migrating focal seizures (EIMFS), West syndrome (WS), early myoclonic encephalopathy (EME), and Dravet syndrome [2].A key feature of these epileptic syndromes is the observation of seizures resistant to standard antiepileptic drugs [1].
These patients with EE could be of interest to pediatric neurologists, since early diagnosis would facilitate treatment planning for patients with EOEE. In the past, seizure onset, electroencephalography (EEG) findings, and neurological features were assessed to diagnose epileptic encephalopathies [1]. Recently, utilization of multi-gene panels via next-generation sequencing (NGS) technology has gained acceleration in order to detect and identify possible genetic variants due to enlighten genetic etiology of heterogeneous epileptic phenotypes [7,8].
The present study focused on the diagnostic yields of targeted multi-gene panels for patients with clinically suspected EOEE in a single-tertiary epilepsy center in Turkey. We aimed to investigate the characteristics of EOEE using the epilepsy multi-gene panel. In addition, we sought to summarize the genetic variations that are frequently found in patients with EOEE as well as their clinical features and treatments.
The patients who were admitted to our hospital with seizure history whose clinical and laboratory findings indicated EOEE and whose etiology was thought to be genetic factors between 2017 and 2020 were included in the prospective study. All patients enrolled to this study were met the following criteria: seizure onset ≤ 24 months of age; progressive developmental deterioration or refractory seizures and clinically suspected EOEE; slow and disorganized background with multiple epileptiform discharges on EEG; no metabolic abnormalities (urine organic acids and plasma amino acids test); and no abnormalities detected on previous genetic tests (chromosome test or chromosomal microarray).
Peripheral blood samples of patients were collected. Genomic DNA was extracted via an automated DNA isolation system (QIASymphony, Qiagen, Germany) using 200 µL from the collected 2 mL peripheral blood samples. Concentration measurement and quality controls of isolated genomic DNAs were performed using a fluorometric measuring device (Qubit 3.0, ThermoFisher, USA). Firstly, the fragmentation process was carried out from the samples and the genomic DNAs were fragmented. Adapter ligation was performed by marking the DNA fragments with specific molecular barcodes. After magnetic bead-mediated purification, target enrichment was performed via multiplex PCR with primers specific to exons and exon-intron junctions of genes in the multi-gene NGS panel designed for EE consisting of KCNQ2, MDH2, NECAP1, PCDH19, PIGA, PLCB1, PNKP, SCN1A, SCN1B, SCN2A, SCN8A, SCN9A, SIK1, SLC12A5, SLC13A5, SLC25A12, SLC35A2, SPTAN1, ST3GAL3, STXBP1, SYNJ1, SZT2, TBC1D24, UBA5, WWOX, AARS, ALG13, AP3B2, ARHGEF9, ARV1, ARX, CACNA1A, CAD, CDKL5, DNM1, DOCK7, EEF1A2, FGF12, FRRS1L, GABRA1, GABRB1, GABRB3, GNAO1, GRIN2B, GRIN2D, GUF1, HCN1, HNRNPU, ITPA, KCNA2 and KCNB1 genes.
Purification process specific to the target regions was repeated and tamplicons were then prepared for library generation by adding universal molecular barcodes. After the bead-mediated final purification process, the library pool was created by qPCR quality and quantity determination from the sample libraries. The library pool was loaded into MiSeq (Illumina, USA) platform and sequenced.
Three-stage quality controls were performed after sequencing: sequence quality control, quality assesments of variants, bioinformatics analysis of variants. As part of the sequence quality control, the total amount of yield and the number of reads passing filter; Within the scope of variant quality controls, the read depth, variant type, variant frequency, number of forward/reverse reads, position of the variant on the gene and quality score of the detected variants were evaluated.
Finally, bioinformatics analyzes were performed to evaluate the clinical significance of the variants in line with the data obtained. In this context, international databanks (Human Genome Mutation Database (HGMD), NCBI dbSNP database, PubMed and disease-specific sources), and the pathogenicity classification of the variants was made in accordance with the ACMG guideline. Possible impacts of novel or clinically unknown variantswere evaluated via in-silico analysis tools (SIFT, B-SIFT, Polyphen-2, MutationTaster, BLOSUM, PROVEAN, CADD, DANN, GeneSplicer, PhyloP, MaxEntScan and QCI Inferred Activation etc.).
Statistical analyses were performed using SPSS version 21.0 (IBM Corporation, NY, USA). Descriptive statistics were used and expressed as median (range). Chi-squared test was used to evaluate the differes between groups; p < 0.05 was considered statistically significant.
Ethics committee approval for this study was obtained from xxxx University Medical Faculty Ethical Committee with year 2019 and numbered 84. Informed consent was obtained from the families of all patients.
Thirty-seven patients with clinical and laboratory findings who were considered to have EOEE were included in the study. Among them, 16 (43.3%) were male and 21 (56.7%) were female. The mean age of seizure onset of the patients was 3.5 months, and the onset of the first seizure ranged from day 1 to 24 months. There was consanguinity between the parents of 19 (51.4%) patients. The demographic characteristics of the patients are summarized in table 1.

Table 1: The demographic findings of the patients (n:37).
WS: West Syndrome; EME: Early Myoclonic Encephalopathy; OS:
Ohtahara Syndrome
The data obtained from the targeted multi-gene panel studied with the NGS method were defined with various software, segregation analyzes and in silico prediction tools. No mutation could be detected in 10 (27%) of 37 patients. Pathogenic variant was detected in five (13.5%) patients. Likely pathogenic was detected in five patients (13.5%), and variant (VUS) of uncertain pathogenicity in seventeen patients (46%). Classification remained unchanged in all reanalysed VUS variants. In total, the causative variant (pathogenic, likely pathogenic and VUS) was detected in 17 patients. Mutation of unknown clinical significance was found in 10 patients.
Seventeen different causal variants were found in 13 different genes in the patients included in the study. The most common causal variant was detected as three VUS variants in the CACNA1A gene. Mutations of unknown clinical significance were found in 10 patients. Some patients had mutations of unknown clinical significance in more than one gene. The distribution of mutations detected in the patients is shown in table 2, and the clinical features of the patients with causal variants are shown in table 3. In this prospective study, the diagnostic value of targeted multi-gene panel analysis in EOEE patients was found to be 27% (P,OP) and 45.9% (P,OP and VUS). The calculated diagnostic values are shown in table 4.
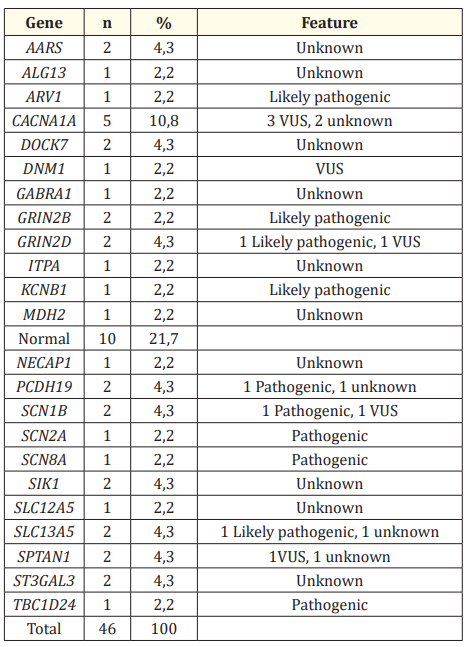
Table 2: Distribution of detected genetic mutations.
Table 3: Mutations and clinical characteristics of the patients
EEG: Electroencephalography, EME: Early Myoclonic Encephalopathy, OS: Ohtahara Syndrome,
WS: West Syndrome

Table 4: Diagnostic values obtained by targeted gene panel analyzes in the patients.
Early-onset epileptic encephalopathy is a severe form of epilepsy, which affects children in early infancy and causes devastating neurological disorders. They have variable genetic heterogeneity. Etiology is diverse in EOEE, which can progress with different clinical findings [1]. In cases where acquired causes cannot be detected, the investigation of genetic etiology is gaining importance both to understand the pathogenesis and to develop rational and specific treatments for pathogenesis. Recently, multi-gene panels with NGS technology have been used to discover possible genetic variants responsible for EOEE’s [7,8]. In a recent study, while the diagnosis rate was reported as 15.4% with targeted single gene sequencing, this rate increased to 46.2% with the use of epilepsy multi-gene panels [9]. In another study, with the use of targeted NGS panels for epileptic encephalopathies, the diagnostic yield in patients increased from 10% to over 25% [10].
In our study, pathogenic in five patients, possibly pathogenic in five patients, and VUS variant in seven patients were determined. In this study, the diagnostic value of pathogenic and possibly pathogenic variants was determined as 27%, and the diagnostic value of all variants (P, OP, VUS) was determined as 45.9%. In a total of 13 different genes, 17 different causative variants were identified. The genes in which variants were detected in our study are CACNA1A, GRIN2D, GRIN2B, PCDH19, SCN1B, SCN2A, SCN8A, KCNB1, SLC13A5, TBC1D24, ARV1, DNM1 and SPTAN1.
Voltage-gated sodium channels, which consist of one major α-subunit and one or more β-subunits, have been shown to be associated with the conduction of action potentials in the brain [11]. The best-known member of this group is the SCN1A gene, which has hundreds of mutations in epilepsy since it was first discovered in 2000 [12]. Sodium voltage-gated channel alpha subunit 8 (SCN8A) mutation causing EOEE type 13 comprises 1% of patients of epileptic encephalopathy [13]. SCN8A-encephalopathies are autosomal dominant disorders, which is caused by de novo gain-of-function missense mutations in the gene. This gene is part of the voltage gated sodium channels (VGSCs) gene family. VGSCs plays important role in controlling neuronal excitability. They are primarily expressed in the central nervous system and mutation causes epilepsy [14]. The patients present with drug resistant seizure between age 3 and 7 months (mean age, 5 months; range, 1 day to 18 months). The seizure has no definite pattern but is often provoked. The common seizures are epileptic spasm, tonic, atonic, myoclonic, focal and absence seizure [15]. Literature has suggested that patients with SCN8A encephalopathy respond to the sodium channel blockers such as phenytoin, valproate, carbamazepine, lacosomide, lamotrigine, rufinamide and oxcarbazepine [15]. Fatema., et al. [16] reported that the clinical and genetic features of two cases of SCN8A encephalopathy, presented with intractable epilepsy and developmental delay. In our study, the patient in whom we detected SCN8A mutation had a seizure onset age of four months, had tonic spasm seizures, and his EEG was in burst suppression pattern. The patient’s seizures were resistant to multiple AEDs. The patient had motor developmental delay and intellectual disability. She was treated with phenobarbitone, levetiracetam, vigabatrine and sodium valproate, but seizure was not controlled. Later he was treated with oxcarbazepine, which caused the 75% control of the seizure.
SCN1B encodes for β1, an immunoglobulinlike molecule that modulates the function of voltage-gated sodium (NaV) channels, a family of nine membrane proteins responsible for initiating andpropagating action potentials [17]. As such, mutations in β1, as well as NaV channels, have been linked to epilepsy syndromes [18]. In humans, inherited heterozygous SCN1B variants have been associated with mild-to-moderate epileptic disorders within the genetic epilepsy with febrile seizures plus (GEFS+) spectrum. In contrast, biallelic variants cause EOEE 52, characterized by infantile onset refractory seizures followed by cognitive decline and neurological features such as hypotonia, spasticity, and ataxia [19]. Only a few individuals with EOEE 52 have been reported so far, and a disease causing mechanism remains unclear. Scala., et al. [20] reported identified nine patients from four unrelated families harboring three biallelic variants in SCN1B. All subjects presented with early infantile EOEE 52, a rare, severe developmental and epileptic encephalopathy featuring infantile onset refractory seizures followed by developmental stagnation or regression. The seizures of the patient in whom we identified a pathogenic mutation started when he was four months old boy, his seizures were in the form of tonic spasm, and there was a burst suppression pattern in his EEG in our study.
Mutations in the SCN2A gene, which encodes the α2 subunit of the neuronal sodium channel, have been identified in association with a number of encephalopathy phenotypes, ranging from benign familial neonatal-infantile seizure to more severe forms of epileptic encephalopathy [21]. Tian., et al. [22] presented the case of an infant with a de novo SCN2A mutation with EOEE who had medically refractory seizures that improved with a ketogenic diet (KD) implemented at an age less than 2 months. On the day of his birth, the infant presented with a pattern of convulsions with dozens of episodes per day. The seizures previously were not controlled with successive therapy with phenobarbital, topiramate, and levetiracetam. The infant’s seizures decreased significantly with a combination of KD and medication. In our study, a pathogenic mutation in the SCN2A gene was detected in a patient with a pre-diagnosis of WS. The patient’s seizures started at the 45th day of life. The patient had infantile spasm seizures and EEG finding was hypsarrhythmia. In addition, the patient had developmental delay. Corpus callosum hypogenesis was detected in the patient’s brain neuroimaging.
PCDH19 encodes protocadherin 19 on chromosome Xq22.3. This 1,148-amino-acid protein, highly expressed during brain development, could play significant roles in neuronal migration or establishment of synaptic connections. PCDH19 is composed of six exons, with a large first exon encoding the entire extracellular domain of the protein [23]. Heterozygous PCDH19 mutations were initially identified in epilepsy and mental retardation limited to females, a familial disorder with a singular mode of inheritance as only heterozygous females are affected, whereas hemizygous males are asymptomatic [24]. Recently, mutations in PCDH19, mostly occurring de novo, were shown to be a frequent cause of sporadic infantile-onset epileptic encephalopathy in females [25]. PCDH19 mutations were also identified in epileptic females without cognitive impairment. Typical features of this new epileptic syndrome include generalized or focal seizures highly sensitive to fever, and brief seizures occurring in clusters, repeating during several days [24]. In a study in which PCDH19 gene mutation was screened among 73 SCN1A negative patients (45 girls and 28 boys) with EOEE resembling Dravet syndrome, mutations in the PCDH19 gene were detected in 13 patients. In this study, the mean age of seizure onset in patients with mutations was 9.5 months (between 7.5 and 12 months), and most of the patients had seizures triggered or worsened by fever. The seizure types of the patients were mostly generalized tonic, clonic or tonic-clonic and/or focal seizures with or without secondary generalization [26]. Our patient, in whom a pathogenic mutation was detected in the PCDH19 gene, was female, in line with the literature. The patient’s seizures were observed for the first time at the age of 1 year. His seizures started as focal and generalized tonic-clonic seizures were added to the table in the follow-up. The patient’s EEG was consistent with multifocal epileptiform activity. The patient had motor developmental delay, but in line with the literature, our patient had language development.
The TBC1 Domain Family Member 24 (TBC1D24) encodes a neuronally expressed GTPase activating protein that interacts with small GTPases and regulates endosomal trafficking of synaptic vesicles [27]. Recent exome sequencing studies have revealed that loss of function variants in TBC1D24 cause a wide spectrum of neurodevelopmental disorders, referred to as TBC1D24-related disorders, including nonsyndromic hearing loss, familial infantile myoclonic epilepsy, early infantile epileptic encephalopathy, and DOOR (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures) syndrome which are inherited in an autosomal recessive manner [28]. Nakashima., et al. [29] described a case with early-onset epileptic encephalopathy, in whom exome sequencing detected a novel pathogenic homozygous variant in TBC1D24. She showed severe developmental delay, congenital sensorineural hearing loss and seizures, but the combination of a high dose phenobarbital and potassium bromide was very effective for the seizures. The seizures of the case in which we found a mutation in the TBC1D24 gene started in the neonatal period and the seizures were myoclonic in character. Multifocal epileptiform activity was detected in the patient’s EEG examination. The patient’s seizures were resistant to multiple AEDs. The patient had developmental delay.
In our stduy, we further detected possible pathogen mutations in the ARV1, GRIN2D, SLC13A5, KCNB1 and GRIN2B genes in five patients. Among these possible pathogenic genes, GRIN2D, KCNB1 and GRIN2B are among the genes playing a role in the ion channel. In the literature, it has been reported that ion channel genes (such as SCN1A, SCN2A, SCN8A, SCN9A, SCN1B, CACNA1A, KCNB1, GRIN2B, GRIN2D) play a prominent role in the development of EOEE and more than 600 variants have been described to date30. In a study involving 175 patients with EOEE, genes encoding ion channels (26/56) were found to constitute the largest portion of the mutations detected [30]. In another study, ion channel genes (8/24) were shown to constitute the most common mutations [31]. In our study, it was observed that ion channel genes accounted for 58.8% (10/17) of the causative mutations. This result indicates that dysfunction of ion channels plays a critical role in the pathogenesis of EOEE. In addition, the rate of ion channel genes in causative mutations was higher compared to other studies. It was thought that this might be due to the prevalence of consanguineous marriages in our region.
In our study, VUS variants were detected in 5 genes in 7 patients. VUS variants were found in the CACNA1A, SCN1B, GRIN2D, DNM1 and SPTAN1 genes. VUS variants were detected in 19% of the patients. In the literature, these variants have been associated with the mutation being relatively less damaging and having a milder phenotype and incomplete penetration in the patient. In other studies, VUS variants could not be correlated with genotype-phenotype due to insufficient clinical information, insufficient bioinformatics information, lack of functional data. and it was evaluated to be associated with incomplete penetrance. It is predicted that the rate of VUS detected in our study is due to reasons such as lack of gene-function studies, lack of information in the databases, and the absence of other affected family members from the study.
Targeted epilepsy multi-gene panels have been used more and more recently in clinical studies and research. Various studies on targeted epileptic encephalopathy panels containing 35-265 genes have been reported in the literature, and the diagnostic values of these studies range from 10% to 48.5% [32].
In conclusion, the variability in the diagnostic values reported in the literature can be explained by the different phenotypic spectra of the selected patients, the number of genes in the panels, the reliability of the sequencing device, and the use of different software and databases for the bioinformatics analysis steps. The diagnostic value determined in our thesis study is similar to the literature. This was associated with the correct management of processes such as selection of patients referred for the multi-gene panel, sequencing of target genes, variant filtering, and association of variant with phenotype.
The authors have declared that there is no conflict of interest.
Ethical approval was obtained for this study from the Ethics Committee of Non-Interventional Clinical Trials of the Faculty of Medicine of Cukurova University with the decision year 2019 and numbered 84.
The study was funded by Cukurov University BAP Coordination Unit with the project number: TTU-2019-11508.
Copyright: © 2024 Faruk Incecik., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.