Srilatha Kadali1,2, Shaik Mohammad Naushad2 and Vijaya Lakshmi Bodiga1*
1 Department of Clinical Biochemistry and Molecular Biology, Institute of Genetics and Hospital for Genetics Diseases, Osmania University, Begumpet, Hyderabad, India
2 Department of Biochemical Genetics, YODA Lifeline Diagnostics Pvt. Ltd, Ameerpet, Hyderabad, India
*Corresponding Author: Vijaya Lakshmi Bodiga, Department of Clinical Biochemistry and Molecular Biology, Institute of Genetics and Hospital for Genetics Diseases, Osmania University, Begumpet, Hyderabad, India
Received: November 13, 2023; Published: November 27, 2023
Citation: Vijaya Lakshmi Bodiga., et al. “Phenotyping and Provisional Diagnosis of Mucopolysaccharidoses Based on Machine Learning”. Acta Scientific Paediatrics 6.10 (2023): 33-43.
In view of significant overlapping clinical features in mucopolysaccharidoses (MPS) subtypes, clinicians face difficulty in differential diagnosis, thus requiring the need for a machine learning-based clinical tool for the provisional diagnosis of MPS subtypes. Out of 520 patients with suspicion of MPS, 296 patients were identified with MPS types. We considered 53 clinical symptoms of MPS patients (n = 255) for the differential diagnosis to derive the model. The diagnosis was based on specific enzyme assays. Among mucopolysaccharidoses, MPS I was the most common disease in our study. Different machine learning tools were tested, out of which classification and regression tree (CART) was more promising. The overall prediction showed 79.92% accuracy in determining the subtype of MPS with a precision of 89.31%. Phenotype-based provisional diagnosis of MPS can be emerging as a useful effective tool for clinicians, thus eliminating the need to perform a whole panel of enzymes. Improved therapeutic efficacy can be attained through early diagnosis and specific intervention. Additionally, this study estimated the prevalence of MPS disorders and established the disease-specific cut-offs of enzyme activity that can distinguish affected from carriers.
Keywords: Mucopolysaccharidoses; Clinical Phenotype; Prenatal Diagnosis; Provisional Diagnosis; Machine Learning.
MPS: Mucopolysaccharidoses; LSDs: Lysosomal Storage Disorders; GAGs: Glycosaminoglycans; DMB: Dimethyl Methylene Blue Method; 2-DE: 2-Dimensional Electrophoresis; DBS: dried blood specimen; LC-MS/MS: Liquid Chromatography-Mass Spectrometry; PCPNDT: Pre-Conception and Pre-Natal Diagnostic Techniques; ANOVA: Analysis of Variance; CART: Classification and Regression Tree; AF: Amniotic Fluid; CVS: Chorionic Villi Sample
Mucopolysaccharidoses (MPS) are a group of inherited lysosomal storage disorders (LSDs) caused by the deficiency of specific lysosomal enzymes, which leads to the accumulation of glycosaminoglycans (GAGs) in the body [1]. The clinical manifestations in these patients include coarse facial features, hepatosplenomegaly, short stature, spinal deformities, skeletal dysplasia, dysostosis multiplex, joint contractures, corneal clouding, hernia, and recurrent respiratory infections [2]. To date, 13 MPS diseases have been identified in humans [3].
The current screening methods available to detect MPS disorders are urinary GAG estimation by the dimethylmethylene blue method (DMB) and cellulose acetate 2-dimensional electrophoresis (2-DE) [4]. This urine analysis is followed by advanced enzyme assays that are specific and save time and money. Monitoring GAG levels also has the benefit of tracking treatment progress. In the postnatal period, measuring enzyme levels in leukocytes or fibroblasts is considered the gold standard, while results from dried blood specimen (DBS) need to be confirmed. For prenatal investigations, chorionic villi and amniotic fluid are the preferred samples [5]. With the development of powerful tools such as next-generation sequencing and tandem mass spectrometry, our understanding of disease mechanisms and pathophysiology has improved. Liquid chromatography-mass spectrometry (LC‒MS/MS) has been used to quantify GAGs in different matrices, e.g., urine, plasma, cerebrospinal fluid, and amniotic fluids [6]. Whole exome sequencing is an emerging technique allowing simultaneous screening of several MPS-related genes due to their overlapping clinical features.
GAG estimation by the DMB method is nonspecific in the differential diagnosis, yet this technique is widely used in laboratories as a preliminary test. 2-DE is a qualitative test that is a tedious procedure and time consuming, and interpretation is subjective. Quantitative LC‒MS/MS analysis results in specificity and sensitivity, but the usage of this technique in diagnostics in India has not yet started. Although specific enzyme assays are the gold standard methods for diagnosing MPS disorders, they are expensive, and very few laboratories are performing them. Currently, whole exome sequencing is recommended to screen for many disorders, but the test is expensive, and if we obtain any variant of uncertain significance, we need to depend on a specific enzyme assay for further confirmation of enzyme deficiency.
It is difficult to differentiate MPS types and subtypes due to overlapping clinical presentation [7]. Hence, there is a need to narrow clinical suspicion, as it is time consuming and more cost involved in performing individual enzyme assays, and the turnaround time is very high in cases of whole exome sequencing results. Hence, it is necessary to narrow the diagnosis at the clinical suspicion stage to achieve early confirmatory diagnosis and further early intervention. To narrow down the diagnosis clinically, we proposed an algorithm based on machine learning to differentiate MPS types and subtypes with 255 MPS patient samples confirmed by enzyme analysis. Additionally, the current study aims to estimate the prevalence of MPS disorders across India and to establish the disease-specific enzyme activity cut-off points to distinguish affected from carriers.
Blood samples (n = 520) from patients with suspicion of MPS disorders were received to perform enzyme analysis with an age range of 0.1 - 21 (6.0 ± 6.8) years. Out of 520 urine samples, 380 samples showed elevated GAG levels. The elevated samples were further analyzed by enzyme analysis. Confirmation of the diagnosis of specific MPS types was performed by specific enzyme assays in peripheral blood leukocytes.
Amniotic fluid was collected from 17 prenatal cases where the index patient was diagnosed with MPS disorder. For prenatal screening and diagnosis, amniotic fluid was collected. Amniotic fluid supernatant was used for GAG estimation, and amniotic culture was performed to perform specific enzyme assays from amniocytes.
This study complied with the ethical principles outlined in the Declaration of Helsinki. Informed written consent was obtained from patients/guardians along with detailed clinical history during their enrollment in the study. Amniocentesis was carried out by a certified fetal medicine specialist after obtaining consent as per the Pre-Conception and Pre-Natal Diagnostic Techniques (PCPNDT) act.
Urine samples were collected from 520 patients with suspicion of MPS disorders. To quantify urinary GAGs, we used the DMB method, which does not require initial precipitation of GAGs. We measured optical density at 520 nm and then calculated the DMB ratio by dividing urinary creatinine by the amount of GAG (mg/L), and the units were expressed as mg/mM creatinine [4].
Sodium heparin blood was collected from 380 patients to isolate peripheral blood leukocytes. Tip sonification was used to prepare leukocyte homogenates. Protein estimation was performed using the bicinchoninic acid method. Specific enzyme activities were further estimated, i.e., a-iduronidase, iduronate 2-sulfatase, heparan sulfaminidase, alpha-N-acetylglucosaminidase, N-acetyl glucosamine transferase, N-acetyl galactosamine-6-sulfate sulfatase, galactose-6-sulfatase, b-galactosidase, and b-glucuronidase, using artificial fluorogenic substrates bound to the fluorochrome 4-methylumbelliferone, which was measured at an excitation wavelength of 362 nm and an emission wavelength of 448 nm. A photometric method using para-nitrocatechol sulfate K2 as a substrate was used to estimate arylsulphatase B enzyme activity. 8 The absorbance of para-nitrocatechol was measured at a wavelength of 515 nm.
The amniotic fluid (AF) samples were centrifuged at 1200 rpm for 10 minutes at room temperature. After centrifugation, the supernatant was removed by leaving 2 ml of amniotic supernatant in the tube. To the pellet, 2 ml of culture media (Amniomax II complete media, Gibco) was added, and the cultures were incubated at 37°C in a 5% CO2 incubator.9 Once we reached the needed cell confluency, the cells were harvested further for enzyme analysis. The supernatant was used for GAG estimation.
Fisher’s exact test was performed using a 2×2 contingency table to calculate the performance characteristics of the model (http:// www.statp ages.org, http://www.wessa.net). Analysis of variance (ANOVA) was used to compare variances across the means of different groups.
Classification and regression tree (CART) construction followed by smart pruning was performed using machine learning tool (https://bigml.com/). The apex of this decision tree indicates the most significant clinical features that help in the differential diagnosis of MPS, while further branching indicates additional features to arrive at a provisional diagnosis.
Out of 520 patients, 296 MPS patients were identified. Among 296 MPS patients, 219 (73.9%) were male samples, and 77 (26.1%) were female samples, with consanguinity in 126 (42.5%) patient samples. The age ranged from 1 month to 22 years, with a mean age of 7.66 years.
Our analysis showed that MPS I is the most common disorder, with 67 patients (48 males and 19 females). That is, nearly 23% of the 296 MPS patients we identified. Coming in a close second is MPS II, with 63 male patients (21.3%). MPS IVA was the third most common disease, with 58 patients (37 males and 21 females).
MPS III subtypes (n = 77), including MPS IIIA (n = 26), MPS IIIB (n = 36), MPS IIIC (n = 12), and MPS IIID (n = 3), presented frequencies of 8.8%, 12.2%, 4.1%, and 1.0%, respectively. MPS VI (n = 24) patients, including 14 males and 10 females with a frequency of 8.1%, and MPS IVB (n = 4) patients, including 3 males and 1 female with a frequency of 1.4%, were identified. MPS VII (n = 3) patients, including 2 males and 1 female, were identified with a frequency of 1.0% (Figure 1).
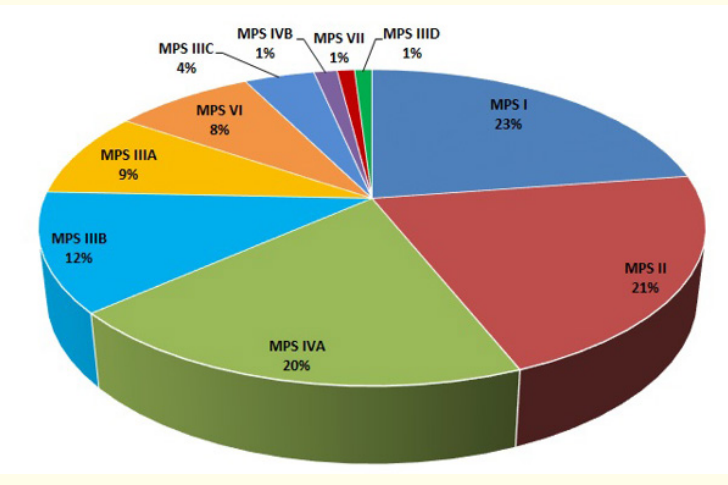
Figure 1: Occurrence of MPS disorders.
Out of 380 samples with elevated GAG levels, 296 samples were identified with deficiency of specific MPS enzymes. GAG estimation by the DMB method showed 77.9% positivity (296/380). We performed enzyme analysis of 10 enzymes for the confirmation of MPS diagnosis. The highest GAG levels were recorded in MPS I, followed by MPS II.
We established specific enzyme activity thresholds that are diagnostic for each MPS type. In MPS 1, alpha iduronidase activity was <6% of the mean normal in all the affected cases, while carriers showed activity between 30 and 73.7%. In MPS II, affected individuals showed <2% mean normal activity, while carriers showed 15-55% mean normal activity. In MPS IIIA, the mean normal activity in affected individuals was <2%, while in carriers, it showed 3454% activity. In MPS IIIB, the mean normal activity was <1% in the affected and 22-43% in the carriers. In MPS IIIC, the mean normal activity was <2% in the affected and 15 -25% in the carriers. In MPS IIID, the mean normal activity was <1% in the affected and 2638% in the carriers. In MPS IVA, the mean normal activity was <1% in the affected and 22-30% in the carriers. In MPS IVB, the mean normal activity was <2% in the affected and 48-92% in the carriers. In MPS VI, the mean normal activity of affected individuals was <4%, while carriers showed 25-41% activity. In MPS VII, the mean normal activity of the affected was <1%, while carriers showed 1626% activity. ANOVA showed that these specific enzyme assays can clearly distinguish controls, affected cases and carriers. In MPS IVB alone, the carrier range overlaps with that of controls (Table 1).

Table 1: Demographic and biochemical study of 296 children affected with different Mucopolysaccharidoses in India.
We considered 53 clinical symptoms for the differential diagnosis and used already diagnosed cases of MPS (n = 255) to derive the model. The diagnosis was based on specific enzyme assays (fluorometric). Different machine learning tools were tested, out of which classification and regression trees (CART) were more promising.
Out of the 53 clinical symptoms, coarse facial features, aggressiveness, mental retardation, hyperactivity, hepatosplenomegaly, behavioral problems, seizures, umbilical hernia, dementia and diarrhea were recognized as the most important determinants of subtypes. MPS II, I, and VII patients presented with coarse facial features as predominant. Aggressiveness is the hallmark feature of all MPS III subtypes. Mental retardation was observed only in MPS I and II patients. Hyperactivity and behavioral problems were identified in patients with MPS IIIA, IIIB, and IIIC. As we have only one case of MPS IIID, we are unable to conclude an association of hyperactivity with MPS IIID. Hepatomegaly is predominant in MPS II and MPS IIIB. Seizures are predominant in MPS IIIC.
We derived a differential diagnostic algorithm using machine learning tools by considering 53 clinical phenotypes of MPS patients as input variables. Ten phenotypes, namely, coarse facies, aggressiveness, mental retardation, hyperactivity, hepatosplenomegaly, behavioral problems, seizures, umbilical hernia, dementia and diarrhea, were identified as the key contributors to the differential diagnosis of MPS. Any patient with a suspicion of MPS disorder having coarse facies, hepatosplenomegaly, and dysostosis multiplex was categorized into MPS I, II, VI, and VII. Along with these features, the presence or absence of mental retardation was differentiated into MPS I and II from MPS VI and VII. Further MPS II and I were classified using corneal clouding and joint stiffness presence or absence, respectively. The presence or absence of corneal clouding and short stature will differentiate MPS VI and MPS VII, respectively.
Aggressiveness, hyperactivity and behavioral problems were seen only in MPS III subtypes. The presence or absence of coarse hair and diarrhea distinguishes MPS IIIA and IIIB from IIIC and IIID. Macrocephaly and hearing loss were observed in MPS IIIA, and in MPS IIIB, they were not observed. The presence or absence of hernia is the key determinant that differentiates MPS IIIC from MPS IIID.
In MPS IV patients, spondyloepiphyseal dysplasia was observed, and MPS IVA and MPS IVB were classified based on the presence or absence of sleep apnea and hearing loss, respectively (Figure 2).

Figure 2: CART model for differential diagnosis of MPS subtypes based on clinical phenotype.
The overall prediction showed 79.92% accuracy in determining the subtype of MPS with a precision of 89.31%. The model performance was excellent for MPS I, MPS III (A, B, C and D), MPS IVA and MPS VII. Model performance is moderate in MPS II, MPS IV B and MPS VI (Table 2). Except for MPS II and MPS IVB, the provisional diagnosis shows a greater degree of concordance with the confirmed diagnosis in all other subtypes.

Table 2: Performance characteristics of the CART model in MPS types based on clinical phenotype.
The mean GAG level was 30.9 μg/ml, with a cutoff of 50.3 μg/ ml in control amniotic fluid supernatant (n = 25) collected at 1621 weeks of gestation. Out of the 17 pregnancies classified as high risk for MPS, GAG analysis seemed to agree with the enzyme assays. Three fetuses were then diagnosed with MPS based on GAG levels - 185 μg/mL (MPS I), 228 μg/mL (MPS II) and 410 μg/mL (MPS VII) with mean and SD of 274 ± 119 ug/mL. Meanwhile, the other 14 pregnancies showed no sign of MPS via GAGs (24.4 ± 10.3 ug/ mL) and respective enzyme assays, and all outcomes ended up being normal (Table 3). Parental blood samples were analyzed in all cases and showed subnormal activities suggesting carrier status.
Machine learning tools are emerging as useful tools for clinicians in diagnosis and therapeutic management. Face2Gene is one such computer-assisted pattern recognition platform that was effective in the provisional diagnosis of known genetic diseases such as mucopolysaccharidoses, Noonan syndrome, 22q11.2 deletion syndrome, etc., based on morphological changes in the face [10]. Earlier, we proposed a machine learning-based algorithm for the differential diagnosis of MPS based on the glycosaminoglycan profile, which showed 96.3% accuracy [11]. Since MPS disorders are heterogeneous with overlapping clinical features, it is difficult to differentiate MPS types and subtypes based on the clinical features of the patients alone. Diagnosis by biochemical and molecular analysis is time consuming. The current study is customized to meet the needs of clinicians and genetic counsellors in establishing provisional diagnosis based on clinical features, thus ordering limited specific enzyme assays to arrive at a definitive diagnosis of MPS subtype. The clinical spectrum observed in our MPS patients corroborated the findings of Kubaski., et al. 2020 [12].

Table 3: List of patients with prenatal testing of MPS cases ($: affected; #: carrier).
The likely higher incidence of MPS in southern India, due to the large population and frequent consanguineous marriages, prompted this study to determine the prevalence of illnesses among children at high risk.
Our study showed MPS I as the most commonly occurring type. The second most common type is MPS II. Our results corroborate those of Verma., et al. 2012, Sheth., et al. 2014 and Gupta., et al. 2018, whose results showed MPS I as a commonly diagnosed MPS disorder in India [13-15]. This differs from Kadali., et al.’s study from India, who found that MPS IVA was the most common type [16]. (19) We also see this to be true in countries such as Egypt[17] (20), Pakistan [18] (21), Tunisia [19], and Denmark and Norway [20]. However, in Turkey [21], Germany, the Netherlands and Emirates [22], MPS III appears to be the most commonly found subtype. Additionally, MPS II has been more common in Brazil [23], Taiwan [24], Malaysia [25], China and Japan [26,27], whereas Canada [28], Colombia [29]. and Mexico’s [30] reports indicate that MPS IVA is the primary form, while Saudi Arab’s statistics show that MPS VI is the initial condition detected.
In Mexico and Tunisia, the estimated combined incidence of MPS was 2.23 [31]and 2.27 [19] per 100,000 live births, respectively. In Taiwan, the estimated incidence of MPS types was 2.04 per 100,000 live births [24], and successful implementation of newborn screening country-wide has led to increased lifespans for patients there compared to elsewhere. Japan reported 1.53 per 100,000 live births between 1982 and 2009, and in Switzerland, the estimated incidence was 1.56 per 100,000 live births, with MPS II being a commonly occurring type [27]. The Czech Republic and Denmark recorded an MPS prevalence of 3.7 [31] per 100,000 live births, while Denmark, Norway, and Sweden recorded 1.77, 3.08, and 1.75 per 100,000 live births [20], respectively. Puckett’s calculation of 10 years of data from 1995 to 2005 in the US found an incidence of MPS types of 1.2 per 100,000 live births [32]. Last, the Netherlands had 4.5 [33] per 100,000 live births.
We only identified four cases of MPS IVB, and many cases of MPS IVB are misdiagnosed as GM1-gangliosidosis since the responsible enzyme (beta-galactosidase) is the same for both conditions. A separate study conducted in the Czech Republic showed that out of 394 affected patients, only one case was reported to be MPS IVB. Additionally, the relative incidence of MPS IVA was observed to be higher than that of MPS IVB, which is consistent with other studies [34].
Our prenatal diagnosis findings are correlated with an earlier study from India using the GAG DMB method for GAG estimation in amniotic fluid [35]. With the advent of mass spectrometry methods, prenatal diagnosis of MPS types becomes possible quantitatively in amniotic fluid with less turnaround time, resulting in 5 affected MPS VI cases [36].
Working groups in countries such as Australia, Portugal and the Czech Republic have been established to raise awareness among medical professionals, and patient support groups also exist to advocate for those affected and their families [31,37]. In India, ICMR has recently created a special task force on lysosomal storage diseases with a focus on raising awareness among clinicians through regional training programmes, gauging the magnitude of these disorders throughout India and establishing common mutation spectra for different LSDs.
Early diagnosis by mandatory nationwide newborn screening programs of MPS via multiplex tandem mass spectrometry method on DBS sample is necessary, as this would provide improved accuracy of the incidence of MPS disorders in any country [38].
Whenever the phenotype is determined with suspicion of MPS and with elevation in GAG, the specific enzyme activity is measured by demonstrating an enzyme assay with fluorogenic or chromogenic substrates. To specify the performance of a specific MPS enzyme assay by the clinician, a reliable screening approach is very important and necessary for MPS determination. Here, the proposed CART model is helpful to clinicians for the differential screening of MPS patients phenotypically. This phenotype-based provisional diagnosis will help clinicians establish a confirmatory diagnosis by examining one or two enzymes rather than performing a whole panel of ten enzymes.
Our study revealed MPS I as the most common MPS type, followed by MPS II. Reliable screening and awareness programs are required to understand more about their true epidemiology. Enzymatic analysis of AF or chrorionic villi sample (CVS) in storage diseases such as MPS is highly reliable for prenatal testing in cases where the diagnosis is confirmed in the index case. Additionally, identifying mutations can aid in early and accurate diagnosis in families at risk. GAG estimation in AF is a beneficial tool for detecting the most common types of MPS, especially when enzyme assay results are inconclusive or AF cultures fail. It is an effective and fast tool that is useful in advanced pregnancies and if an index case diagnosis is unavailable. Disease specific enzyme activity cut-off points established can be used for distinguishing affected vs carriers. Ultimately, this study shows that MPSs are common in South India and that appropriate genetic counseling with prenatal diagnosis during subsequent pregnancies could help reduce the burden of such diseases.
We acknowledge the children and families who participated in the study. We thank Mr. Rajeev Sindhi, MD, Sandor Speciality Diagnostics Pvt. Ltd. for giving permission to carry a part of the study.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Srilatha Kadali: Conceptualization, Methodology, Data curation, Manuscript writing. Shaik Mohammad Naushad: Manuscript revision. Vijaya Lakshmi Bodiga: Conceptualization, Methodology, Supervision. All authors revised and approved the final manuscript.
Copyright: © 2023 Vijaya Lakshmi Bodiga., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.