Aisha A. SehariGammodi1*, Nadia L Hatem2, Maha Y Kamal2, Doreen N Younan3, Shimaa A Shaltout4 and Ashraf M Ayad4
1 pediatrics Department, Faculty of of Medicine, University of Tripoli, Libya
2 Department of Pediatrics, Faculty of Medicine, Alexandria University, Egypt
3 Department of Clinical Pathology, Faculty of Medicine, Alexandria University, Egypt
4 Pediatric Department - Damnhour Teaching Hospital, Egypt
*Corresponding Author: Ashraf M. Ayad , pediatrics Department, Faculty of of Medicine, University of Tripoli, Libya.
Received: April 17, 2023; Published: June 03, 2023
Citation: Ashraf M. Ayad., et al. “Study of Association of Hepcidin Promotor C-582 A>G Variant and Iron Over Load in B-Thalassemia Major”. Acta Scientific Paediatrics 6.7 (2023): 03-10.
Background: Hepcidin, a 25 amino acid peptide expressed in the liver, has been shown to have a major role in iron metabolism controlling iron release from reticuloendothelial and intestinal epithelial cells. Studies reported a potential role of iron loading modifier (Hepcidin) in poly-transfused beta-thalassemic patients for the -582 A>G polymorphic change (rs10421768)2 in the 5' flanking region of the HAMP (Hepcidin AntiMicrobial Peptide) gene encoding hepcidin.
Objective: In this study we studied the genetic variant in the hepcidin gene (HAMP) promotor (C.-582 A>G) and the associations between this variant and iron overload in patients with B-thalassemia major.
Subjects: Two groups were included in this study: Group I: included fifty one children with B-thalassemia major diagnosed by clinical signs, complete blood picture and Hemoglobin electrophoresis Nearly all children in group I were receiving regular red cell transfusion and regular oral iron chelation therapy (Deferasirox)and were clinically followed up in the hematology clinics of Alexandria University Children's Hospital (AUCH) and Damnhour Teaching Hospital. Group II: included twenty-two healthy children of matching age and sex with the previous group as controls.
Methods: All patients included in the study were subjected to thorough history taking with special emphasis on : age at starting transfusion therapy, frequency of packed red cells transfusion, adherence to chelation by oral chelator (deferasirox), thorough clinical examination with special emphasis on: thalassemic features, hepatic and splenic size, cardiac examination, laboratory investigations including: routine investigations to diagnose B- thalassemia i.e, complete blood count, Haemoglobin electrophoresis, liver function tests: Alanine transaminase (ALT) and Aspartate transaminase (AST), kidney function tests: 3300d urea nitrogen (BUN) and Serum creatinine, c -reactive protein (CRP), iron 7rofile {serum iron,total iron binding capacity (TIBC) and serum ferritin (SF)} by ELISA, Genomic DNA was extracted from EDTA whole 7 lood samples using QIAamp DNA blood mini kit 50.C_582A>G (rs 10421768) SNP was detected by 5' nuclease allele discrimination assay using real-time PCR.
Results: The mean serum ferritin was not significantly higher in patients with genotype AG/GG than patients with genotype AA (mean ± SD 2049.23 ± 848.27and 1081.67 ± 120.03 respectively ng/ml),
Conclusion: The c.-582A > G HAMP promoter variant is associated with high serum iron and ferritin levels but with no significant difference between genotypes AG/GG and AA in patients with ẞ-thalassemia major.
Keywords: ẞ-Thalassemia major (TM); DNA; EDTA
ẞ-Thalassemia major (TM) is a hereditary hemolytic anemia res3lting from the reduction or complete absence of ẞ-globin chain synthesis. Thus leads to reduced adult hemoglobin ([HbA] (ɑ2ß2heterotetramer) and excess a-globin content in erythroid cells, in turn resulting in ineffective erythropoiesis and apoptosis in the erythroid lineage. Most ẞ- thalassemia patients therefore require lifelong clinical management by PRBCS transfusion and chelation therapy, with a few having the option of curative but potentially hazardous transplantation of hematopoietic stem and progenitor cells (HSPCs) instead. Clinical presentation of TM occu4s between 6 and 24 months. Affected infants fail to thrive and become progressively pale. Progressive enlargement of the abdomen caused by spleen and liver enlargement may occurr [1].
Chronic in overload represents a serious complication of repeated blood transfusions. Indeed, organ f34ure due to chronic iron overload represents the major cause of death in patients with TM. In addition, ineffective erythropoiesis enhances gastrointestinal iron absorption and recycling of iron from macrophages and reticuloendothelial system through down regulation of a newly discovered peptide hormone hepcidin. Iron overload in thalassemia can be mainly assessed by serum ferritin, liver iron concentration (LIC), cardiac T21 MRI) and superconducting quantum interference device (SQUID) [2].
Complications of iron overload include: cardiac complications, hepatic complication and endocrine complications as hypogonadism, delayed puperty, growth retardation, diabetes mellitus, hypothyroidism, hypoparathyroidism and osteoporosis [3].
The body iron metabolism is based on a highly efficient system of iron preservation and recycling by which only up to a 10th of the daily need is replaced by duodenal absorption from diet. Excess iron is either not absorbed, or is retained in enterocytes. After approximately 2 days these cells are shed from the tips of the villi into the luminal intestinal contents. This apparently regulated iron uptake and the absence of a physiological excretion mechanism to release excessive iron in case of iron overload [4].
In 2000, hepcidin was initially isolated from plasma ultrafiltrate and named liver- expressed antimicrobial peptide (LEAP-1). The development of severe iron overload by knocking out the gene in mice has suggested that hepcidin is involved in iron metabolism, whereas this key element in regulation was underlined by the discovery of hepcidin mutations in patients [5].
Hepcidin, encoded by HAMP gene, is a recently discovered 25 amino acid peptide that, in addition to being involved in innate immunity (15) appears to play a crucial role in iron metabolism in humans, regulating both iron absorption from the intestine and its recycling by macrophages [6].
Moreover, hepcidin regulates cellular iron export into plasma. When hepcidin concentrations are low, ferroportin (Fpn) molecules are dislodged on the plasma membrane and export iron. When hepcidin concentrations increase, hepcidin binds to ferroportin molecules and induces their internalization and degradation, and iron release is decreased progressively [7].
Regulated by different stimuli, which act as positive or negative controllers.
There are four main active regulation pathways (erythroid, iron store, inflammatory and hypoxia mediated regulation) that control hepcidin production through different signaling pathways. These pathways must be closely attached to match iron supply to erythropoietic demand and, in turn, to maintain adequate plasma iron concentrations [8].
After hepcidin is secreted into the circulation by the hepatocytes it eventually exits the body in the urine. Its effect on the decrease in serum iron levels in mice appeared to take place within 4 hours in a dose-dependent way that was sustained for more than 48 hours [9].
Several methods for hepcidin quantification in human serum and urine have been reported. There are two antibody-based techniques, a dotblot assay, which has been used to measure hepcidin in urine and is considered semi-quantitative [10], and a commercially available ELISA assay measuring serum prohepcidin.
As hepcidin deficiency causes iron overload in iron loading anemias such as B- thalassemia, agents that can mimic hepcidin function or potentiate its endogenous production would be expected to prevent systemic accumulation of iron.
Several mutations located in the hepcidin gene (HAMP) have already been associated to the development of iron overload or hereditary hemochromatosis (HH). Additionally, some HAMP promotor were described, however their functional sequences remain unclear. One of them is the poly morphism C.-582A>G (AG,GG and AA) that was recently described as worsening iron overload phenotype in beta- thalassemia major Study of these polymorphisms of HAMP promoter gene seems warranted now.
The aim of the work was to identify the genetic variant (C._582 A>G) in the hepcidin gene (HAMP) promotor and to assess the associations between these variant and iron status parameters.
This study enrolled two groups of children:
Genomic DNA was extracted from EDTA whole blood samples using QIAamp D7 A blood mini kit 50 (QIAGEN, Germany, cat. no. 51104). C_.582A>G (rs 10421768) SNP was detected by 5‘ nuclease allele discrimination assay using real-time PCR.
The last three laboratory investigations and CBC were done for both thalassemic cases and controls.
Data were fed to the computer and analyzed using IBM SPSS software package version 20.0. Comparisons between groups for categorical variables were assessed using Chi-square test and Fisher‘s Exact or Monte Carlo correction. Student t-test was used to compare two 290ups for normally distributed quantitative variables. Significance of the obtained results was judged at the 5% level.
There was no significant statistical difference betwee22 the age in ẞ- thalassemia major patients and the age of control children (p = 0.062). Also, t35e was no significant statistical difference between the two groups regarding sex (p = 0.700) using Chi square test.
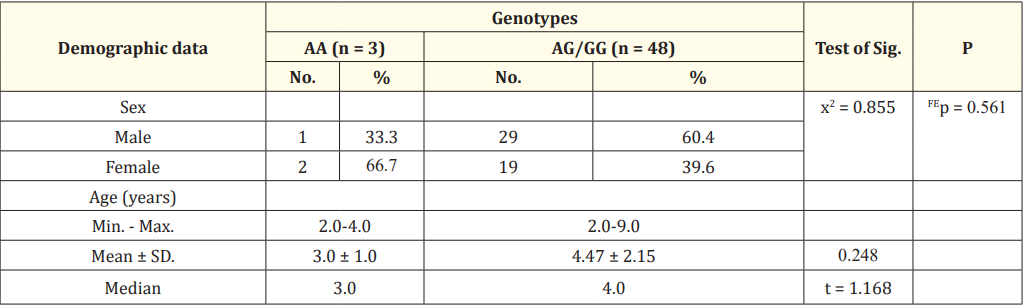
Table 1: Relation between genotypes and demographic data in the studied thalassemic patients.
x2: Chi square test FE: Fisher Exact for chi square test.
t: Student t-test.
Table 1 show relation between genotypes and demographic data in studied thalassemic patients.
Regarding sex, there is no significant relation between sex and genotype in the studied thalassemic patients (p = 0.561) using Fisher Exact for Chi square test. Also, there is no statistically significant relation between age and genotype (p = 184).

Table 2: Relation between genotypes and Haemoglobin level in the studied thalassemic patients.
t: Student t-test.
Table 2 show that there is insignificant statistical difference between HAMP genotypes in studied TM patients regarding haemoglobin level.
However, the mean haemoglobin level in the studied thalassemic patients was significantly lower than that of controls (p < 0.001).

Table 3: Relation between genotypes and iron profile in the studied thalassemic patients .
t: Student t-test.
Table 3 show the relation between genotypes and serum ferritin level in the studied thalassemic patients. The mean serum ferritin level in the studied thalassemic patients with the genotype AG/ GG 2049.23 ± 848.27 ng/ml was insignificantly higher than that in the studied thalassemic patients with the genotype AA 1081.67 ± 120.03 ng/ml (p = 0.056).
However, the mean serum ferritin level in 31 studied thalassemic patients was significantly higher than that of the controls with (p < 0.001).
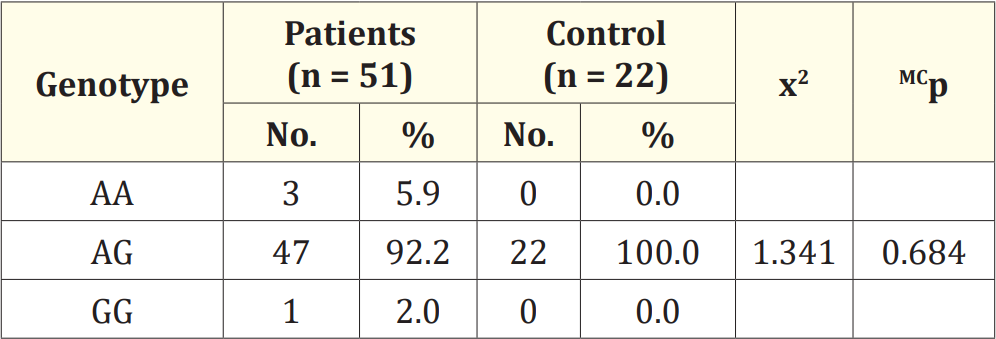
Table 4: Comparison between the two studied groups according to genotype.
x2: Chi square test for comparing between the two studied groups
MC: Monte Carlo for chi square test
Table 4 show that 92.2% of B-thalassemia major patients have the hepcidin promotor c._582 A>G, 5.9% have the hepcidin promotor c._582 A>A and 2% have the hepcidin promotor c._582 G>G. On the other hand 100% of 19e controls have the hepcidin promotor c.- 582 A>G with no significant statistical difference between the two groups using Monte Carlo for Chi Square test.

Table 5: Relation between genotypes and presence of thalassemic features in patients group.
x2: Chi square test.
MC: Monte Carlo for chi square test
Table 5 show relation between genotypes and thalassemic features in the studied thalassemic patients. In the patients with genotype AG/GG, 35.4% did not have thalassemic features, 64.6% have thalassemic features. While, the patients with genotype AA all do not have thalassemic features. Using Monte Carlo for Chi Square test, there is no statistically significant relation between genotypes and thalassemic features (p = 0.111).

Table 6: Relation between genotypes and Hb electrophoresis in the studied thalassemic patients.
t: Student t-test.
Table 6 show that the comparison of results of Hb electrophoresis (Hb F, A1 A2) among different HAMP genotypes of thalassemic patients was statistically insignificant.

Table 7: Relation between genotypes with age at starting transfusion therapy and frequency transfusion in the studied thalassemic patients.
t: Student t-test.
Table 7 show that different HAMP genotypes don‘t significantly affect age at starting transfusion therapy or frequency of transfusion.
TM is the most severe form of ẞ-thalassemia which presents during the first months of life (5) and is cha 20 terized by the multiple effects of tissue hypoxia due to chronic hemolytic anemia and ineffective erythropoiesis (IE) requiring regular red blood cell transfusion to sustain life. Transfusion therapy leads to excess iron accumulation which deposits in most organs resulting in tissue damage. Scoring criteria of severity of thalassemia includes HB at steady state (g/dl), Age at receiving first blood transfusion (year), size of spleen, age at thalassemia presentation (year) and growth and development.
The classic clinical picture of thalassemia major is currently only seen in some developing countries, 18 which the resources for carrying out long-term transfusion programs are not available. The skeletal changes include deformities of the long bones of the legs and typical craniofacial changes (bossing of the skull, prominent malar eminence, depression of the bridge of the nose, tendency to a mongoloid slant of the eye, and hypertrophy of the maxillae, which tends to expose the upper teeth).
As the body has no effective means for removing iron, the only way to remove excess iron is to use iron binders (chelators), which allow iron excretion through the urine and/or stool. Asa general rule, patients should start iron chelation treatment once they have had 10-20 transfusions or when ferritin levels rise above 1000 ng/ ml.
Multiple studies have shown deferasirox is effective in decreasing cardiac iron burden; however, the results regarding if it is effective in removing cardiac iron in cases of severe iron overload are conflicting.
Hepcidin is the main regulator of iron homeostasis and, therefore, a better understanding of its regulation is of great interest. In the present work, conducted on 51 BTM patients and another 22 controls we identified the hepcidin promotor C.-582 A>G variant and the possibility of its association with iron overload.
Regarding sex, 30 patients are males and 21 patients are females. Their age ranged from 2-9 years.14 out of 22 controls are males and 8 are females, the age of these controls ranged from 3-5 years. So, all the study participants were comparable regarding age and sex.
Regarding iro 18 rofile, the mean serum iron and serum ferritin levels in the studied thalassemic patients were significantly higher than that of the controls with (p < 0.001).
This goes with Aboul-Enein., et al. (2015) who studied peripheral expression of hepcidin gene in Egyptian ẞ- thalassemia major. This study included 50 TM patients and 20 healthy volunteers as acontrol group. The serum ferritin level of these TM patients was high (mean ± SD 3759.9 ± 2165.9 ng/ml) [11].
In the present study, real time PCR revealed that 47 of the studied ẞ-thalassemia major patients have the hepcidin promotor c._582 A>G, 3 patients have the hepcidin promotor c._582 A>A and one patient have the hepcidin promotor c._582 G>G.
On the other hand, all controls have the hepcidin promotor c.- 582 A>G with no significant statistical difference between the two groups.
Also, our results showed that the 2 groups of the studied thalassemic patients with genotype AG/GG and patients with genotype AA were comparable as regards the age and also were comparable as regards the sex distribution ratio as the female precentage was 39.6% and 66.7% respectively and p > 0.05.
The current study showed that by comparing HAMP genotypes regarding thalassemic features, hemoglobin electrophoresis, age at starting transfusion therapy, and frequency of transfusion there was insignificant difference between these genotypes.
Hence, the results of the present study showed that either the presence of allele G in c.- 582 A > G variant in thalassemic children (n = 48) or the presence of allele A (n = 3) did not have a significant relation with iron profile parameters as serum iron or serum ferritin. This may be attributed to the small number of cases in each group. Also, there was no significant correlation with Hb level in each group.
These results are in agreement with Bruno., et al. who did not find any association between the c._582 A>G genotype and serum iron, serum transferrin, transferrin saturation or ferritin level, which might reflect no differences in liver iron concentration [13].
According to Andreani., et al. the c.-582 A>G HAMP-P variant did not show an impact on the level of iron loading in the regular chelated patients [13], while it influenced the LIC values and the serum ferritin (SF) among the patients receiving an irregular chelation treatment. These observations suggest that this HAMPP polymorphism may be associated with different functionality of the HAMP promoter. The c.-582 A>G HAMP-P substitution might, therefore, represent a risk factor for thalassemic patients by promoting iron overload in these subjects. On the other hand his results showed that this effect can be overcome by a regular and appropriate chelation therapy. Other factors, such as age and variable duration of transfusion regimens, might account for the wide range of LIC and SF levels observed in both wild type (WT) and HAMP-P variant patients. The reason why in some HAMP-P individuals iron overload was comparable to that of wild-type patients could be due to the fact that they are heterozygous for the SNP (Single Nucleotide Polymorphism) and, therefore, show an incomplete penetrance or compensatory mechanisms.
These data together with the fact that HAMP is regulated by many different signals suggested that the effect of the c.-582A > G polymorphism on hepcidin levels, and therefore on the iron status in-vivo is most likely negligible. These authors hypothesized that the c- 582A > G variant in the human HAMP promoter would have no effect on the hepcidin transcription in normal situations but it might have some effect in physiopathological situations where more hepcidin was needed. However, further studies would be necessary to confirm this hypothesis.
Recently; a new hormone, erythroferrone (ERFE), had been identified. It mediates hepcidin suppression during stress erythropoiesis, ERFE is produced by erythroblasts in response to erythropoietin. ERFE expression is greatly increased in thalassemic mice models, which contributes to the suppression of hepcidin (Kautz., et al. 2014) [14].
Copyright: © 2023 Ashraf M. Ayad., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.