Diana Reis Monteiro*, Liliana Sá, Daniela Pinto and Gustavo Rocha
Department of Neonatology, Centro Hospitalar Universitário de São João, Porto, Portugal and Department of Pediatric/Neonatology, Centro Hospitalar Entre o Douro e Vouga, Santa Maria da Feira, Portugal
*Corresponding Author: Diana Reis Monteiro, Department of Neonatology, Centro Hospitalar Universitário de São João, Porto, Portugal and Department of Pediatric/Neonatology, Centro Hospitalar Entre o Douro e Vouga, Santa Maria da Feira, Portugal.
Received: August 16, 2022; Published: October 12, 2022
Citation: Diana Reis Monteiro., et al. “Clinical Approach to Multiple Congenital Renal Cysts”. Acta Scientific Paediatrics 5.11 (2022): 07-11.
Renal cystic diseases are a diverse group of diseases that can manifest in utero, infancy, or throughout childhood and adulthood. They can appear as an isolated finding or as part of a syndrome and can affect one or both kidneys. Differential diagnosis often remains a challenge since numerous diseases can cause cystic kidney malformations and imaging patterns change over time.
We report a case of a 1-month-old female infant that was observed in a neonatology consultation for multiple renal cysts on the left kidney, first described in the prenatal ultrasound. Our patient was a full-term neonate, born by vaginal delivery, with a birth weight registered of 2865gr. The physical examination at the first appointment showed no abnormal findings. The first ultrasound evaluation performed after birth confirmed a left kidney with a small amount of undifferentiated renal parenchyma and several simple cysts, the largest with 43x28mm, suggestive of multicystic dysplastic kidney. The right kidney was described as normal. Laboratory tests showed normal values of hemoglobin, renal function and electrolytes. Additional investigation with abdominal and transfontanelar ultrasounds revealed no other malformations. Follow-up with serial ultrasonographic evaluation showed gradual reduction of the left kidney size as well as a growing right kidney showing compensatory enlargement.
From genetic syndromes to some non-hereditary forms of polycystic kidney diseases, in situations like the case presented, it is mandatory to consider all causes of multiple unilateral cysts with perinatal presentation. Recognition of such an extensive variety of renal cystic diseases is essential for early detection and start of treatment if needed. Although considered a benign condition, multicystic dysplastic kidney requires long-term follow-up to ensure that the contralateral kidney undertakes appropriate compensatory hypertrophy and to identify other associated anomalies. Routine monitoring should include serial ultrasound evaluation as well as blood pressure measurement, urinalysis, and kidney function studies, particularly in children who present contralateral abnormalities that are at greater risk for developing chronic kidney disease.
Keywords: Unilateral Multicystic Dysplastic Kidney; Infants; Cystic Kidney Diseases.
ADPKD: Autosomal Dominant Polycystic Kidney Disease; ARPKD: Autosomal Recessive Polycystic Kidney Disease
Renal cystic diseases are a diverse group of diseases that can manifest in utero, infancy, or throughout childhood and adulthood [1,2]. Most of them are due to nonhereditary fetal malformations or genetic disorders [3,4]. They can appear as an isolated finding or as part of a syndrome, [3,5] and can affect one or both kidneys [1,2]. Recognition of such an extensive variety of neonatal renal cystic diseases is important to include all options in the differential diagnosis, which is essential for early detection of the right diagnosis and start of treatment if needed [5].
We report a case of a 1-month-old female observed in a neonatology consultation for multiple renal cysts, first described in the prenatal ultrasound. Our patient was a full-term neonate, born by vaginal delivery, with a birth weight registered of 2865gr and Apgar scores 9/10/10. There was no family history of renal disease or known genetic disorders. Physical examination at first appointFigure 1: Renal ultrasound performed on the third day-of-life, showing multiple renal cysts, the largest marked by yellow points in the image, with 43 x 28mm. ment showed no abnormal findings. The first ultrasound evaluation performed after birth confirmed a left kidney with a small amount of undifferentiated renal parenchyma and several simple cysts, the largest with 43 x 28mm, suggestive of multicystic dysplastic kidney, as shown in figure 1. Right kidney was described as normal. Laboratory tests showed normal values of hemoglobin, renal function and electrolytes. Additional investigation with abdominal and transfontanelar ultrasounds revealed no other malformations.
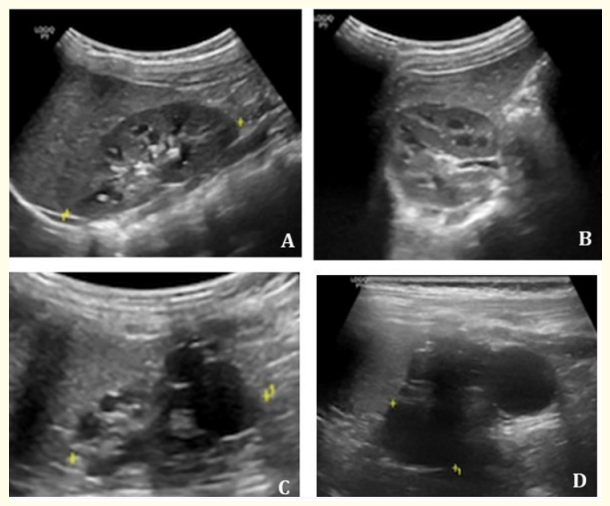
Figure 1: Renal ultrasound performed on the third day-of-life, showing multiple renal cysts, the largest marked by yellow points in the image, with 43 x 28mm.
The patient was observed regularly throughout the next two years. She showed adequate growth patterns, with weight and length both on the 50th centile. Neurodevelopment milestones were achieved at a normal age range. A single episode of a urinary tract infection was diagnosed and treated at nine months, due to Escherichia coli. Serial ultrasonographic evaluation at 3, 6, 12 and 22 months showed gradual reduction of the left kidney size and consequently its cysts, as well as a growing right kidney showing compensatory enlargement with a calculated size greater than the 95th centile, as shown in figure 2.
Multicystic dysplastic kidney consists of numerous noncommunicating cysts separated by dysplastic tissue. [1,3,5-8] It is described as the most common cause of cystic renal disease in children, [1,7] affecting 1 per 1,000-4,300 live births [3,5-7]. Diagnosis is usually made by prenatal ultrasound [3]. Most cases are unilateral, with the left kidney being more often involved, [3,7] and these patients are usually asymptomatic or present with an abdominal mass [1,6,7]. Bilateral disease is uncommon and is associated to a poor prognosis for survival [1,8]. Natural progression without intervention is partial or complete involution of the affected kidney [1,3,4,6,7]. The main reason for long-term follow-up is to ensure that the contralateral kidney goes through an appropriate compensatory hypertrophy and to exclude other anomalies such as vesicoureteral reflux or urinary tract obstruction [4-7]. Since complications like the risk of malignancy, infection or hypertension are rare, treatment is usually conservative [1,3,4,6,7]. Routine monitoring should include serial ultrasound evaluation at 6 months, 2 years, 5 years, 10 years, and 15 years of age, to monitor adequate contralateral kidney growth and affected kidney involution, as well as blood pressure measurement, urinalysis (to detect proteinuria), and kidney function studies, particularly in children who present contralateral abnormalities that are at greater risk for developing chronic kidney disease [3,4,6,8]. Ultrasound evaluation is the basic method for diagnosis and follow-up, nevertheless renal scintigraphy provides additional information about function of renal cortical tissue, the involvement of the contralateral kidney, and presence of any scar, which is why it is suggested to perform one at least once in these patients [7]. Voiding cystourethrogram however, should be reserved for patients with recurrent urinary tract infections or other anomalies detected by ultrasound or scintigraphy [7].
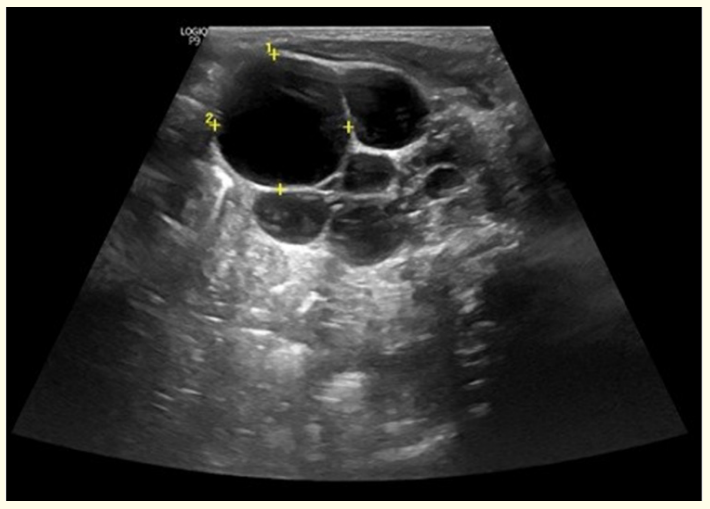
Figure 2: Renal ultrasound performed at 22 months exhibited a growing right kidney with compensatory enlargement (A and B) and a left multicystic dysplastic kidney with size reduction (C and D).
Differential diagnosis often remains a challenge because numerous diseases can cause cystic kidney malformations and imaging patterns evolve over time [2], namely, genetic syndromes such as autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD), Bardet-Biedl syndrome, Beckwith-Wiedemann syndrome, Ivemark syndrome, Jeune syndrome, juvenile nephronophthisis, Von-Hippel-Lindau syndrome, Hajdu-Cheney syndrome, Meckel-Gruber syndrome, orofacial-digital syndrome type 1, or Zellweger cerebrohepatorenal syndrome; and some non-hereditary forms of polycystic kidney diseases like multicystic dysplastic kidney, cystic dysplasia, simple renal cysts, or complex renal cysts. More rarely, renal cysts can be acquired [3,5]. Regarding the case presented it is mandatory to consider other causes of multiple unilateral cysts with perinatal presentation, such as cystic dysplasia, unilateral start of ADPKD and ARPKD. It may be difficult to distinguish ADPKD renal cysts from those caused by ARPKD in newborns [1].
ADPKD, with dominant inheritance, is the most common inherited kidney disease [1,5]. It may be diagnosed at any stage from before birth through childhood, [8] but its most frequent presentation is in adulthood, affecting 1 in 400 to 1,000 individuals [4,5]. It results from a defect in polycystin 1 and 2, encoded by PKD1 in 85% of cases and PKD2 in 15% of cases [1,5]. It is characterized by multiple small cysts of variable size, unevenly distributed along the renal parenchyma on both cortical and medullary levels [4,8]. Clinically, it can cause not only progressive cystic dilation of both kidneys but also manifestations in the gastrointestinal tract, cardiovascular system, reproductive organs, and the brain [1,5,8]. Diagnosis is based on family history and ultrasound findings, however imaging studies are inconclusive in most patients younger than five years [5]. Regarding prognosis, PKD1 is associated with a more severe disease, presenting in utero with the Potter sequence and earlier development of cysts, while PKD2 has a more benign course and later onset [5]. It causes end-stage renal disease in 5-10% of patients [1,5].
ARPKD, with recessive inheritance, usually presents in the neonatal period, as a bilateral enlargement and elongation of the renal collecting system, with large palpable flank masses, causing variable degrees of renal dysfunction [5]. This results from a defect in the fibrocystin/polyduct in protein, encoded by PKHD1, that affects not only the renal system but can also cause hepatic abnormalities, [5,8] such as hepatic fibrosis, bile duct ectasia, and biliary segmental dilatations [4]. Diagnosis is made during pregnancy when prenatal ultrasound reveals symmetric enlarged renal masses, with hyperechogenic kidneys, showing poor or inverted corticomedullary differentiation, microcysts, a small or nonvisualized bladder with no urine, and oligohydramnios [1,5,8]. Regarding prognosis, it is associated with higher infant mortality and amongst the survivors, 50% develop end-stage renal disease by 10 years of age [5].
Cystic kidney dysplasia is characterized by a single or multiple cysts within an abnormal renal parenchyma [3,8] in which the kidney contains primitive ducts and cysts, and nonrenal tissues such as cartilage, fat, and hematopoietic tissue [3]. It is often associated with obstruction of the urinary tract, like posterior urethral valves or ureteropelvic junction obstruction [3,4]. It usually involves the whole kidney but can be localized as segmental dysplasia [3,8].
Renal cystic diseases may be identified by ultrasound prenatally as early as 17 weeks [1]. After birth, diagnosis and follow-up of these patients should not focus only on kidneys, but should include all abdominal organs, since some syndromes that present renal cysts have other abdominal anomalies associated [4]. For children with unilateral multicystic kidney disease and adequate hypertrophy and normal parenchyma of the contralateral kidney, genetic testing in the absence of extrarenal manifestations is not recommended because of the low risk of specific genetic disease and the good prognosis [2].
Although considered a benign condition, an early diagnosis of multicystic dysplastic kidney is crucial for an appropriate management with prompt identification of associated anomalies to preserve contralateral kidney function [1,7].
We have no conflicts of interest to disclose.
Copyright: © 2022 Diana Reis Monteiro., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.