André Costa Azevedo*, Ana Isabel Sequeira, Dalila Rocha, Isabel Soro, Beatriz Sousa, Armando Laranjeira
Neonatal Intensive Care Unit, Pediatrics Department, Unidade Local de Saúde do Alto Minho, Viana do Castelo, Portugal
*Corresponding Author: André Costa Azevedo, Neonatal Intensive Care Unit, Pediatrics Department, Unidade Local de Saúde do Alto Minho, Viana do Castelo, Portugal.
Received: June 08, 2022; Published: June 27, 2022
Citation: André Costa Azevedo., et al. “Primary Ciliary Dyskinesia: An Uncommon Cause of Neonatal Respiratory Distress”. Acta Scientific Paediatrics 5.7 (2022): 46-49.
Primary Ciliary Dyskinesia is a rare and genetically heterogeneous disorder characterized by immobility, dysmotility or absence of cilia, causing chronic oto-sino-pulmonary disease. Approximately 50% of patients have situs inversus totalis. Genetically, the most common genes involved are DNAH5 and DNAI1. Newborns with Primary Ciliary Dyskinesia may have respiratory distress with mild transient tachypnoea or mild hypoxemia, rhinitis or atelectasis, usually associated with poor feeding. Six days after birth, a full-term male newborn was admitted to the paediatric emergency department for intermittent respiratory distress, mainly during breastfeeding, associated with nasal congestion and sporadic productive cough. Physical examination revealed acrocyanosis, jaundice and bilateral basal crackles. Preductal pulse oxygen saturation was 90% on room air requiring supplementary oxygen therapy. Chest X-ray demonstrated dextrocardia. Echocardiography confirmed dextrocardia without structural heart disease. Abdominal ultrasound showed the presence of situs inversus totalis, with no other malformations. During the hospitalization nasal congestion, cough and hypoxemia progressively resolved. Coexistence of total situs inversus and unexplained newborn respiratory distress suggested the presence of Primary Ciliary Dyskinesia. Genetic testing showed two different variants of DNAH5 gene confirming the diagnosis. The infant was referred to a Paediatric Pulmonology centre and Paediatric and Otorhinolaryngology outpatient clinic. He also started chest physiotherapy sessions and immunoprophylaxis against respiratory syncytial virus. Although neonatal presentation is unusual, Primary Ciliary Dyskinesia should be considered for neonates presenting with respiratory distress of unclear cause. Early diagnosis is important in order to decrease damage to the respiratory system from recurrent infections and improve the quality of life and prognosis.
Keywords: Ciliary Motility Disorders; Primary Ciliary Dyskinesia; Newborn; Respiratory Distress; Shortness of Breath.
NICU: Neonatal Intensive Care Unit; PCD: Primary Ciliary Dyskinesia
Respiratory distress is one of the most common causes for admission to Neonatal Intensive Care Unit (NICU) and occurs in up to 7% of newborn infants [1]. This is recognized as any signs of breathing difficulties in the neonate such as tachypnoea, nasal flaring, chest retractions, grunting or cyanosis. Although the causes of respiratory distress in a newborn are diverse, they are usually related to pulmonary pathology [1]. Causes can also include noncyanotic congenital heart disease, anatomical, neuromuscular, haematological, metabolic or infectious pathology and less common, genetic disorders [1]. Due to the broad list of conditions that cause newborn respiratory distress, diagnosis is often challenging.
Primary Ciliary Dyskinesia (PCD) is a rare and genetically heterogeneous disorder characterized by immobility, dysmotility or absence of cilia, causing chronic oto-sino-pulmonary disease [2,3]. Prevalence is variable and unknown, but it is thought to be between 1:4000 and 1:40000 [4]. Motile cilia are present on the epithelial cells apical surface, clearing airway mucus, bacteria and debris and it can also be found in the middle ear, paranasal sinuses, lung, female and male reproductive tract and brain ependyma [4]. The anomalous mucociliary clearance caused by the ciliary motion defects leads to recurrent sinorespiratory infections [2].
Ciliary dysfunction may result from a mutation in a large number of genes. With the exception of two genes (X-linked syndromic genes RPGR and OFD1) almost all follow an autosomal recessive inheritance and result from a defect of dynein arms, radial spokes and microtubules structure [3,5]. The most common genes involved are DNAH5 and DNAI1 [3].
The authors present a case of a newborn that 6 days after birth developed a respiratory distress syndrome.
A full-term male newborn was delivered at 38 weeks of gestation by a 27-year-old woman gravida 2 para 1. The neonate was born by normal vaginal delivery with a birth weight of 2900g, appropriate for gestational age and an Apgar score of 10 at 1 and 5 minutes of life. Maternal history was significant for obesity and gestational diabetes managed by dietary changes. The mother had received prenatal care and laboratory tests, including negative group B Streptococcus, and antenatal scans were unremarkable. Parents are non-consanguineous and the first child (a 1-year-old boy) had no significant past medical history. The newborn physical examination was normal and he was discharged at two-days of life. Newborn screening tests (metabolic, pulse oxymetry and hearing) proved to be normal.
Six days after birth he is admitted to the paediatric emergency department for intermittent respiratory distress, mainly during breastfeeding, associated with nasal congestion and sporadic productive cough. The physical examination reveals acrocyanosis, jaundice and bilateral basal crackles. Preductal pulse oxygen saturation is 90% on room air requiring supplementary oxygen therapy. There is no difference in preductal versus postductal saturations nor in blood pressure measurements in the four members. Chest X-ray (Figure 1) demonstrates dextrocardia without peribronchial infiltration or pulmonary consolidation. Laboratory findings show normal white cell-count, with normal C-reactive protein and total bilirubin of 17 mg/dL. Microbiological samples are negative for SARS-CoV2 and other respiratory viruses. He is admitted to the NICU for further management and investigations.
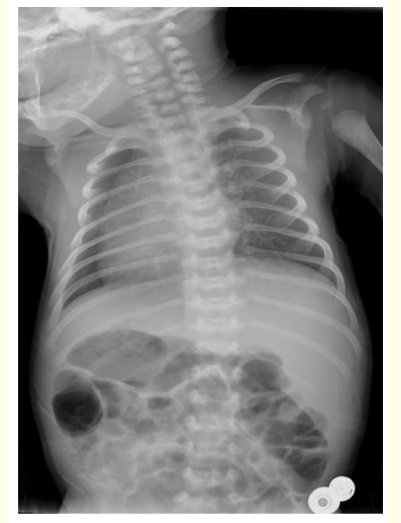
Figure 1: Chest X-ray showing total situs inversus.
During the admission the echocardiography confirms dextrocardia without structural heart disease and the abdominal ultrasound shows the presence of situs inversus totalis, with no other malformations. During the hospitalization nasal congestion and cough progressively resolve. It is observed short desaturation episodes (minimum value of 70%) with spontaneous recovery with maximum amount of oxygen needed of 0,5 liter per minute through nasal cannula. The coexistence of total situs inversus and unexplained newborn respiratory distress points to PCD. Genetic testing was performed, showing two different variants of DNAH5 gene (NM_001369.2: c.4237C > T p. (Gln1413*) and NM_001369.2: c.12397G > T p. (Glu4133*)) confirming the PCD diagnosis.
On day ten of admission, the newborn is discharged without requiring supplemental oxygen. The infant is referred to a Paediatric Pulmonology centre and Paediatric and Otorhinolaryngology outpatient clinic. He also starts chest physiotherapy sessions and immunoprophylaxis against respiratory syncytial virus. As infective complications, the infant develops an acute otitis media and an acute bronchiolitis at one and two-month-old, respectively. Parents are referred to a Genetic physician for genetic counselling for future pregnancies, confirming compound heterozygous mutations.
The entire respiratory tract is affected and despite a full-term gestation, 80% of the newborns with PCD has respiratory distress with mild transient tachypnoea or mild hypoxemia (and may require supplemental oxygen for a few hours to days after birth), rhinitis or atelectasis, usually associated with poor feeding [3,6]. Most of these newborns are asymptomatic immediately after birth, but develop respiratory distress of unknown cause, being hospitalized within the first few weeks of life [3]. In this particular case, although asymptomatic at birth, the hospitalized newborn developed mild respiratory distress of unknown cause, being hospitalized at the sixth day of life.
Persistent nasal obstruction is often noted since the neonatal period and during preschool and school-age daily rhinitis throughout the year associated with sinusitis exacerbations are common [4,6]. During childhood, daily wet cough and repeated episodes of bronchitis and pneumonia leads to the development of bronchiectasis, which is a marker of disease progression [6,7]. Particularly during the first year of life, at least 80% develop recurrent otitis media with chronic effusion [3]. In this case, the infant already had two infectious complications, an acute otitis media and an acute bronchiolitis, during the first 2 months of life.
Many patients may develop conductive hearing difficulties [4]. Approximately 50% of patients have situs inversus totalis which combined with chronic sinusitis and bronchiectasis is named Kartagener Syndrome [2]. Adult males are often infertile due to reduced sperm motility and females have also reduced fertility [3].
Diagnosis of PCD may be delayed and it requires a high level of suspicion because its main symptoms are also frequent in healthy children [2,4]. Measurement of nasal nitric oxide, assessment of ciliary beat frequency and pattern by high-speed video microscopy, analysis of ciliary ultrastructure by electron microscopy and genetic studies are some of the diagnostic tests that can be used [3]. Definitive diagnosis is made when patients with clinical phenotype suggestive of PCD have: a) ciliary ultrastructure defects by electron microscopy or b) non-ambiguous biallelic mutations [8].
In result of the recurrent respiratory infections, these patients are advised to see a pulmonologist 2-4 times per year [3]. Moreover, these patients should also be followed-up by an otorhinolaryngologist 1-2 times per year, for monitoring chronic rhinosinusitis and recurrent acute otitis media [3]. Transtympanic ventilation tubes use are controversial and patients may be warned about multiple insertions, postoperative otorrhea or permanent tympanic perforation [3,4]. There are no current effective therapies targeting the genetic mutation. Prevent therapies include daily airway clearance, nasal sinus lavage and chest physiotherapy. Pneumococcal and annual influenza vaccines and seasonal immunoprophylaxis against respiratory syncytial virus are recommended [3].
PCD should be considered for neonates presenting with respiratory distress of unclear cause. Although the classic triad of symptoms may not be present in the neonatal period, the coexistence of total situs inversus and respiratory distress points to the diagnosis of PCD. Early diagnosis is important in order to decrease damage to the respiratory system from recurrent infections and improve the quality of life and prognosis. This case report highlights the management of neonates with PCD, which includes daily nasal sinus lavage, chest physiotherapy to enhance airway clearance, identification and treatment of airway infections, proper vaccination according to national protocols (including pneumococcal and annual influenza vaccines) and seasonal immunoprophylaxis against respiratory syncytial virus.
Thank you to: Dr. Francisca Martins for the admission and first care of the patient; Dr. Ariana Teles and the pulmonology team from Centro Hospitalar São João, Porto – Portugal, for the immediate follow-up in pediatric pulmonology outpatient clinic; Dr. Armando Laranjeira for the essential assistance in the management of the patient and all the medical and nursing NICU’s team from Unidade Local de Saúde do Alto Minho.
Authors state no conflict of interest.
Copyright: © 2022 André Costa Azevedo., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.