Srushti Gandhi1*, Rashid Merchant2, and Snehal Mallakmir2
1 Department of Paediatrics, Nanavati Super Specialty Hospital, Mumbai, Maharashtra, India
2 Department of Genetics, Nanavati Super Specialty Hospital, Mumbai, Maharashtra, India
*Corresponding Author: Srushti Gandhi, Department of Paediatrics, Nanavati Super Specialty Hospital, Mumbai, Maharashtra, India.
Received: March 04, 2022; Published: April 28, 2022
Citation: Srushti Gandhi., et al. “A Case Report of a Child with Langerhans Cell Histiocytosis”. Acta Scientific Paediatrics 5.5 (2022): 31-36.
Langerhans cell histiocytosis (LCH) is a rare clonal disease of the monocyte-macrophage system, comprising of uncontrolled proliferation of CD1a+/CD207+ dendritic cells (DCs). It has a wide range of symptomatology making early diagnosis and intervention extremely challenging. LCH may range from mild single system involvement with isolated skin or bone lesions to life-threatening multi-system involvement. Furthermore, when risk organs are involved, even with aggressive treatment, 10-year survival rates are only 50%, further emphasizing the need for maintaining a high index of suspicion and early intervention. We present the case of a 2-year-old girl who arrived at our clinic with a painless lump over the head following a trivial head injury, and on further evaluation was found to have histopathologically confirmed multisystemic LCH. Response to treatment has been favourable.
Keywords: Langerhans Cell Histiocytosis; CD1a+/CD207+; Monocyte-Macrophage System; Litterer Siwe Disease; Hand-SchüllerChristian Disease.
LCH: Langerhans Cell Histiocytosis; CD: Cluster of Differentiation
Langerhans cell histiocytosis (LCH) (formerly called “histiocytosis-X”) is a rare proliferative disease of monocyte-macrophage system, with uncontrolled proliferation of CD1a+/CD207+ dendritic cells (DCs) [1]. Its clinical presentation ranges from isolated single system involvement, skin/bone lesions, to life-threatening multi-system involvement. Early diagnosis is challenging due to wide symptomatology, however essential for timely treatment initiation and improved outcomes. Histiocytosis-X included: eosinophilic granuloma, Hand-Schüller-Christian disease and LettererSiwe disease [2-6]. At present, LCH is classified into single-system single site (SS-s), single-system multi-site (SS-m), and a multisystem type (MS) [7-10].
We report a 2-year-old girl, born of non-consanguineous marriage, who came with history of trivial head injury 6-weeks ago followed by swelling in the right supraorbital area. The mother noticed a nontender swelling, without impulse on crying, gradually increasing in size since 1-week (3 x 4 x 1cms) (Figure 1A). Our initial differential diagnoses included growing skull fracture, metastatic skull lesions and Langerhans cell histiocytosis. Active questioning revealed history of intermittent ear discharge since 1.5months of age, for which she had received multiple antibiotics. Ear examination showed a granuloma/polyp like tissue in the external auditory canal (Figure 1B). She had a crusty, scaly, seborrheic dermatitis like rash all over the scalp since 4months of age (Figure 1C) and longitudinal lines on all nails since 1year (Figure 1D, E). Apart from mild hepatomegaly, other findings on examination were within normal limits. We did a CT brain to see the origin of the swelling. It showed expansile lytic lesions involving right frontal bone and within right mastoid air cells (Figure 2A-C). Hence an MRI brain was done, which showed a gourd-shaped soft-tissue mass extending from the subcutaneous to the intracranial space with heterogeneous post contrast enhancement (Figure 2D-F). MRI abdomen showed hepatomegaly (Figure 3C-D). USG abdomen showed mild prominence of intrahepatic biliary radicles (Figure 3A-B). Blood investigations revealed anaemia, thrombocytosis and raised GGTP, alkaline phosphatase and LDH. Due to high index of suspicion for multisystemic LCH, we did a biopsy of the auditory polyp, which showed large polygonal cells with surrounding inflammatory infiltrate composed predominantly of eosinophils. On immunohistochemistry, CD1a and S-100 were positive in many of the large cells (Figure 6A-C). We now had a complete clinical, radiological and histopathological diagnosis of multisystemic LCH. We did a full body PET CT to aid in staging and prognostication which showed metabolically active lytic lesions in right frontal and mastoid bone were seen). Periportal cuffing with mild dilatation of intrahepatic biliary radicles were noteds in the liver. Lung windows showed multiple cysts, with lower lobe predominance (Figure 4A-D, Figure 5A-D). Due to multisystemic involvement with involvement of risk organs, we started her on Stratum 1 of LCH-IV, International Collaborative Treatment Protocol for Children and Adolescents with Langerhans Cell Histiocytosis [14]. Vinblastine and Corticosteroid treatment was started. Clinical improvement was noticed as early as after the first dose of Vinblastine and Corticosteroids itself (Figure 8). Gradually she started showing improved activity, improvement in blood and other biochemistry parameters. The swelling over the supraorbital area reduced. At 6 weeks repeat PET CT has shown complete resolution of right mastoid lesion, with decrease in volume of right frontal bone lesion from 17.4 cm3 to 3.4 cm3 (Figure 7). Overall, she has shown significant clinico-radiological improvement (Figure 9A-B).

Figure 1: A: Swelling in right supra-orbital area. B: A granuloma/polyp like tissue in the external auditory canal. C: Seborrheic dermatitis like rash all over the scalp. D: Longitudinal lines on all finger nails.

Figure 2: A-F: Shows expansile lytic lesions involving right frontal bone (thick arrows) and within right mastoid air cells (arrows). On bony window settings, lytic lesion demonstrates bevelled edge with no sclerotic rim (notched arrow). The right mastoid lesion is seen extending to external auditory canal with no ossicular erosion. MRI demonstrates gourd-shaped soft-tissue mass extending from the subcutaneous to the intracranial space with heterogeneous post contrast enhancement.
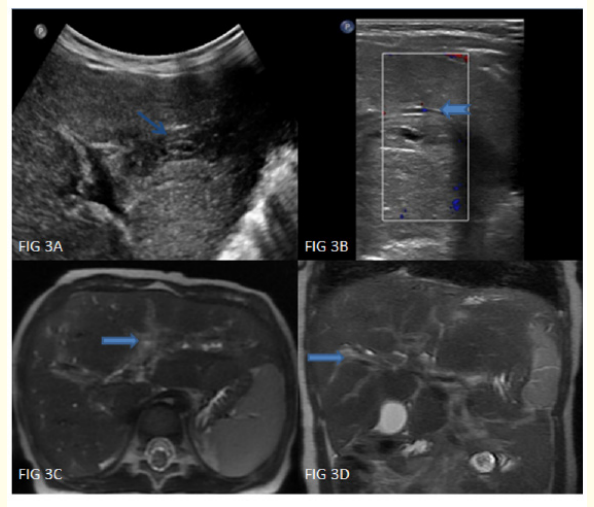
Figure 3: A-D: High resolution ultrasonography showing mild prominence of intrahepatic biliary radicles (notched arrows) with diffuse periportal hypoechogenicity in both lobes of liver (arrows). MRI demonstrates hepatomegaly with diffuse T2 hyperintense periportal cuffing in both lobes of liver (thick arrows).

Figure 4: A-D: Lung window settings show multiple cysts of variable sizes with lower lobe preponderance (thick arrows). Associated thickening of interlobular interstitium seen (arrow).

Figure 5: A-D: On PET-CT, metabolically active lytic lesions in right frontal and mastoid bone seen (arrows). Periportal cuffing in liver is seen, with associated mild dilatation of intrahepatic biliary radicles (notched arrows).
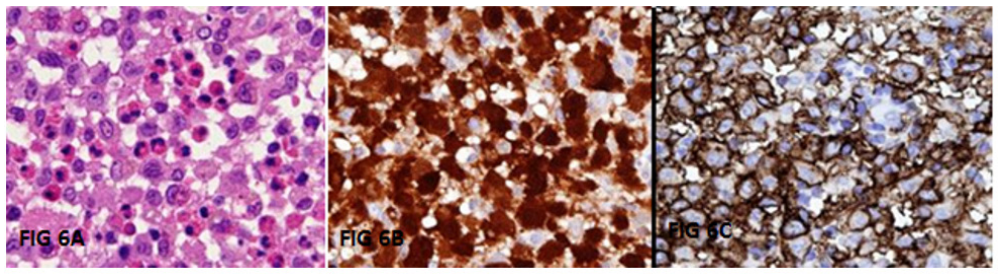
Figure 6: A-C: On histopathological examination, large polygonal cells with round to oval nuclei and indistinct cytoplasm with surrounding inflammatory infiltrate composed of predominantly eosinophils were seen (6A). On Immunohistochemistry (IHC), CD1a and S-100 were positive in many of the large cells (6B and C).

Figure 7: Follow up PET-CT showing near complete resolution of right mastoid lesion with right frontal bone lesion showing decrease in volume from previous of 17.4cm3 to present of 3.4cm3.
Histiocytoses are a group of rare clonal disorders characterized by the accumulation of cells derived from dendritic cells or macrophages. The first classification of histiocytosis, published Formerly, histiocytoses was classified in 1987, into 3 categories: langerhans cell (LC) or non-LC-related, and malignant histiocytoses (MH). A new classification based on phenotype, histology, molecular alterations, and clinical as well as imaging characteristics has evolved over time, currently dividing the histiocytoses into 5 groups [1] (Table 1), LCH being the most common one. It is approximated to affect 4 to 5 per million children, between the ages of 0 to 15 every year. The median age at diagnosis is 3.5 years. Highest incidence is in infancy, before 1 year of age, followed by a decreased incidence thereafter [11]. Unfortunately, despite much research and great advancements, the pathophysiology of LCH remains unconfirmed and is stipulated to be the result of a neoplastic process or an anomalous autoimmune response. LCH also has a wide range of symptomatology, emphasizing the need for having a high index of suspicion. Organ affection in order of commonality is as follows: bone (any bone, excluding hands and feet) (80%), skin (33%), pituitary (25%), liver (15%), spleen (15%), hematopoietic system (15%), lungs (15%), lymph nodes (5-10%), and the central nervous system excluding the pituitary (2-4%). Bony lesions may be unifocal or multifocal. They may be painless, as was the case in our patient, whose presenting symptom was a nontender, painless lump on the head. The second most frequently involved organ system is skin. The lesions may be single or multiple, and localised, circumscribed or diffuse. Identification of risk organ involvement is highly essential for deciding the treatment protocol and prognosis. Risk organs include the hematologic system, the spleen and the liver [2]. The lung is also included by many as a risk organ, however it must be noted, that in the absence of other risk organ involvement, lung lesions in LCH leading to mortality is extremely rare. Several treatment protocols and strategies have been tried for Multisystemic LCH, from minimal therapy to intensive chemotherapy. A randomized trial demonstrated that Vinblastine and etoposide, with a single dose of corticosteroids, were equally effective treatments for multisystem LCH, but those who failed to respond within 6 weeks were at greater risk for treatment failure and need for different treatment [12]. Similarly, our patient was given a treatment consisting of an initial 6-week course of vinblastine and prednisone, and at 6 weeks, there was a net regression of the tissue component. For children above 2 years of age at the time of diagnosis, a 10-year survival rate is reported to be 97%; whereas for those younger than 2 years, the rate is 77% [13]. Overall survival rate is approximately 71% at 10 years. With involvement of risk organ systems (spleen, liver, bone marrow, lungs), this 10-year survival is said to drop to 50% even with aggressive treatment [13].

Figure 8: Reduction is size of supraorbital lesion after 1st dose of Vinblastine + Steroids.
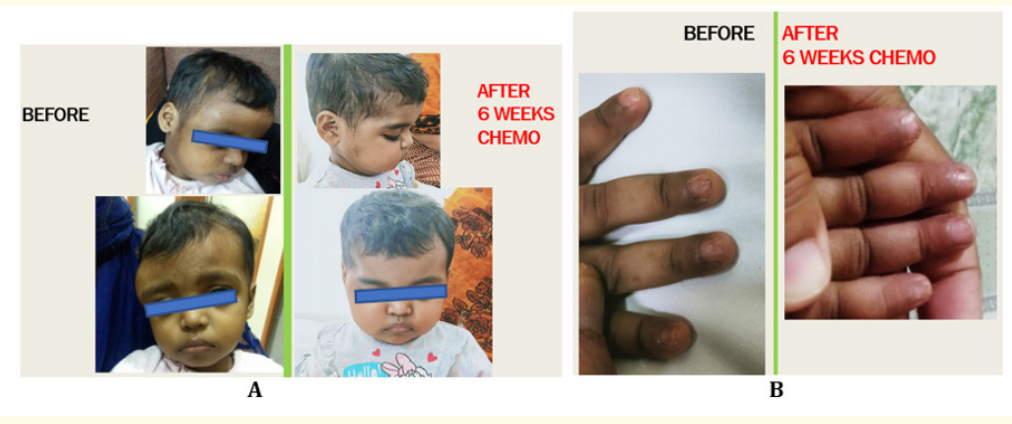
Figure 9: A: Complete clinical resolution of supraorbital lesion after 6 weeks of chemo. B: Improvement of nail changes after 6 weeks of chemo.
LCH is a rare entity with various and non-specific clinical presentations. Bones being the commonest site of involvement, a swelling or lump in the head must raise a suspicion of LCH in a child. Recognition of symptoms and having a high index of suspicion is necessary. This is difficult since there is extreme clinical heterogeneity and these kids present to a variety of specialists (e.g., general paediatricians, dermatologists, dentists and orthopaedic surgeons). The diagnosis of LCH is based on accurate histological and immunophenotypic examination of tissue. Prognosis of multisystemic LCH is poor even with aggressive treatment, making early diagnosis and intervention extremely important. The treatment involves systemic chemotherapy and corticotherapy aiming at prevention of irreversible damage to normal tissues and long-term consequences. It is advised to have active involvement of a paediatric oncologist for proper risk stratification, appropriate treatment plan and long-term follow-up.
Table 1: histiocytoses classification showing the 5 groups of diseases.
Dr. Narayan Jayshankar, ENT Surgeon, for conducting the biopsy of the ear granuloma; Dr. Gita Verma, HOD, Histopathology, for her invaluable expertise in providing a immunohistopathological diagnosis for the child; Dr. Rajashree Rawal, Senior Pathology Consultant, for not only her medical expertise on the histopathology of the biopsy, but also for funding the investigations and treatment of the child and providing moral support to the family; Dr. Mitusha Verma, Dr. Nimish Kamath and team for helping in risk factor assessment and response to treatment via MRI, USG, CT and PET CT; and Dr. Mukesh Desai, Senior Haematoncologist, for his invaluable expertise in the management.
We have no conflict of interest to declare.
Copyright: © 2022 Srushti Gandhi., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.