Srushti Gandhi1*, Snehal Mallakmir2, and Rashid Merchant1
1 Department of Paediatrics, Nanavati Superspecialty Hospital, Mumbai, Maharashtra, India
2 Department of Genetics, Nanavati Superspecialty Hospital, Mumbai, Maharashtra, India
*Corresponding Author: Srushti Gandhi, Department of Paediatrics, Nanavati Superspecialty Hospital, Mumbai, Maharashtra, India.
Received: March 04, 2022; Published: April 28, 2022
Citation: Srushti Gandhi., et al. “Multiple Sulfatase Deficiency: A Clinical Report with Neonatal Presentation”. Acta Scientific Paediatrics 5.5 (2022): 26-30.
Multiple Sulfatase Deficiency is a rare autosomal recessive inborn error of metabolism caused by mutations in the sulfatase modifying factor 1 gene which encodes the formylglycine-generating enzyme, and resulting in accumulation of sulfatides, sphingolipids, sulphated glycosaminoglycans and steroid sulphates in various tissues. More than fifty different mutations and less than hundred cases have been published so far. We report a new case of MSD presenting in the neonatal period with respiratory distress, feeding difficulty, hypotonia, ichthyosis and dysmorphology. This patient was detected with a known homozygous pathogenic variation in SUMF1 gene c.1043C>T (p.A348V).
Keywords: Multiple Sulfatase Deficiency; MSD; SUMF1 Gene; Formylglycine-Generating Enzyme; Ichthyosis.
FGE: Formylglycine-Generating Enzyme; MSD: Multiple Sulfatase Deficiency; IEM: Inborn Error of Metabolism; SUMF1: Sulfatase Modifying Factor 1 Gene; MLD: Metachromatic Leukodystrophy; CT: Computed Tomography.
Multiple sulfatase deficiency (MSD) (OMIM # 272200) is a rare, autosomal recessive, inborn error of metabolism (IEM). Mutation in the sulfatase modifying factor 1 gene (SUMF1) is responsible for the disorder. More than 50 different mutations in the SUMF1 gene have been identified [1,2]. SUMF1 encodes the formylglycinegenerating enzyme (FGE). This enzyme is responsible for the posttranslational modification of a cysteine residue, which is essential for the activity of sulfatases in in the endoplasmic reticulum.
Hence, mutation in this gene results is accumulation of sulfatides, sphingolipids, sulphated glycosaminoglycans and steroid sulphates in various tissues [3]. There are a total of 17 known sulfatases, of which 9 single sulfatase deficiencies have been identified [4,5]. MSD, thus has a more heterogeneous clinical picture and includes a combination of all features seen in these individual 9 single sulfatase deficiencies such as Hunter syndrome, Sanfilippo A syndrome, Morquio syndrome, Maroteaux–Lamy syndrome, metachromatic leukodystrophy (MLD), and X-linked ichthyosis. These include neurological, developmental, skeletal, cardiovascular, ophthalmological, auditory, dermatological, dental and various other organ system involvement. The severity and phenotype of MSD depends on the severity of FGE protein instability and its residual action after the mutation [6,7]. MSD is classified into subtypes according to the age of onset, as early neonatal, late infantile severe (LIS) and late infantile mild (LIM) and rare juvenile (mild) disease.
We report a new case of MSD presenting in the early neonatal period with respiratory distress, feeding difficulty, hypotonia, ichthyosis and dysmorphology. This patient was homozygous for a previously documented SUMF1 mutation c.1043C > T (p.A348V) [8].
An 11-month-old, full term baby girl was brought to our hospital in view of intermittent respiratory distress and inability to accept full oral feeds. The baby had a severely deformed and depressed nasal bridge, midfacial hypoplasia as well as other dysmorphic features such as sutural overriding, sandal gap (Figure 1A, B, C). She was born to nonconsanguineous parents, a primigravida mother via lower segment caesarean section in view of pregnancy induced hypertension and intrauterine growth restriction. The baby was small for gestational age, vigorous at birth, and had cried immediately. However, she developed respiratory distress soon after for which she had required nasotracheal intubation for 3 days. They noticed that the intubation relieved the distress completely. CT scan of the airway showed a depressed nose with small height of the nasal bridge, S shaped deviated nasal septum which closely abutted the lateral wall of the right nasal cavity anteriorly and left middle and superior turbinate with obliteration of the upper part of left posterior nasal cavity (Figure 3A, B, C, D). A chromosomal analysis was done, which revealed normal female genotype. She gradually started accepting full oral feeds and was discharged at 1 month of life. However, at 2 months of age, she had to be re-admitted for intermittent breathing difficulty, history of choking and inability to accept full oral feeds. At the time, a dye study under C-ARM with small volume dye revealed normal swallowing coordination. When larger volumes were given, part of it was found to come out from the oral cavity. There was no evidence of aspiration into airway or gastro-oesophageal reflux. Laryngo-tracheo-bronchoscopy was done which showed features of laryngomalacia. Gavage feeds were started via a nasogastric tube. Gradually, oral feeds were restarted in small quantities. A feeding plan was devised to gradually increase oral feeds, while giving the rest via gavage feeding.
Currently, at arrival at our clinic, the baby’s breathing difficulties had shown improvement. However, the baby continued to require gavage feeds along with minimal oral feeds. She had a global developmental delay with no head holding, no reaching out and no social smile achieved. She was only 3.8kg at 11 months of age, 54cms long and a head circumference of 39cms. All parameters were below the 3rd centile for age. Apart from the afore-mentioned findings, a complete general examination revealed small palpebral fissures, short limbs, posteriorly placed dysplastic pinnae, ape thumb deformity, dry scaly skin and ichthyosis (Figure 2A, B). She had generalised hypotonia, power was normal. On enquiry it was found that the parents feel the baby’s facial and other physical features resembles the mother’s sister’s daughter, who expired in infancy due to breathing difficulties. Xray spine showed hemivertebrae of the cervical and thoracic vertebrae with only 11 pairs of ribs. Butterfly vertebra was noticed at D12 level. A kypho-scoliotic deformity was also noticed in the lower dorsal and upper lumbar region (Figure 4A, B). In view of above findings, a whole exome sequencing was sent. This revealed a homozygous c.1043C > T (p.A348V) likely pathogenic variant in the SUMF1 gene, suggestive of an autosomal recessive condition, Multiple Sulfatase Deficiency (MSD). The child was hence diagnosed with neonatal type MSD. Parents were counselled regarding further evaluation of the child with respect to cardiac, neurological, hearing abnormalities; poor prognosis and future family planning.
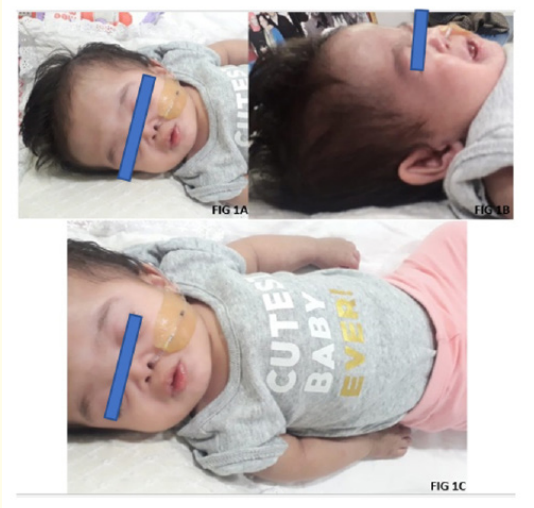
Figure 1: A, B, C: Severely deformed and depressed nasal bridge, Small palpebral fissure, short limbs, posteriorly placed dysplastic pinnae.
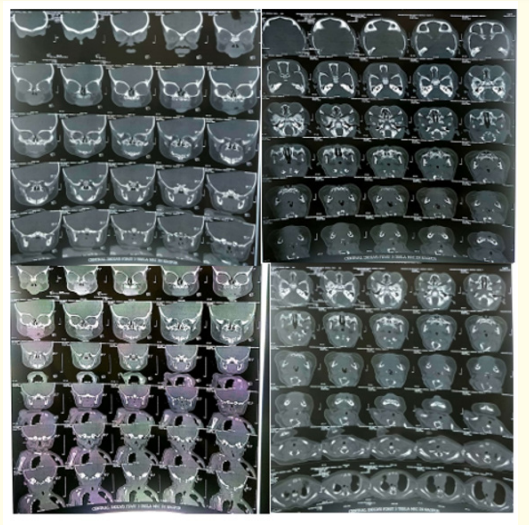
Figure 3: A, B, C, D: CT scan of the airway showed a depressed nose with small height of the nasal bridge, S shaped deviated nasal septum which closely abutted the lateral wall of the right nasal cavity anteriorly and left middle and superior turbinate with obliteration of the upper part of left posterior nasal cavity.

Figure 2: A, B: Dry skin, Ichthyosis.
It is classified according to the age of onset, as early neonatal, late infantile (0 to 2 years, severe or mild), and juvenile (2 to 4 years, mild). Patients may show various phenotypical features like a neurodegenerative course of disease such as that seen in metachromatic leukodystrophy. Organomegaly, gargoyle facies, developmental delay may be seen similar to various mucopolysaccharidoses [9]. Skeletal abnormalities are similar to those found in Chondrodysplasia punctata type 1 and skin changes of X-linked ichthyosis may be present [10]. Earlier the onset, more severe are the symptoms and poorer are the chances of survival while life span may be longer with subacute clinical presentation in juvenile cases. Genotype phenotype corelation was seen in few studies which showed a correlation of both stability and residual activity of FGE variants with the clinical picture of selected MSD patients. Severe impairment of both FGE stability and residual enzyme activity was associated with the most severe clinical phenotype whereas the mildest phenotype was associated with the highest residual FGE activity among the studied variants. But more data is still needed to help establish a clear genotype/phenotype correlation as well as predict the clinical course for the studied SUMF1 mutations. In our case, the flat facies, skeletal changes and ichthyosis all led us to the first differential of Conradi-Hunermann Syndrome (X-linked dominant chondrodysplasia punctata) [11], however the characteristic punctate epiphyseal stippling/calcification was absent. Data suggests that ichthyosis is an important indicator of MSD and the best marker of the disease in neonates. Neonatal onset MSD shows extremely poor survival rates beyond infancy, however, one case report of siblings with the same mutation as our case, demonstrated that they survived well beyond 2 years of age under advanced multidisciplinary team care.

Figure 4: A, B, C: Chest and Abdomen Xray show situs solitus, no major abnormality in the major organs or soft tissues. Xray spine showed hemivertebrae of the cervical and thoracic vertebrae with only 11 pairs of ribs. Butterfly vertebra was noticed at D12 level. A kypho-scoliotic deformity was also noticed in the lower dorsal and upper lumbar region.
Documentation of low activity levels in a minimum of two sulfatase enzymes and/or genetic analysis demonstrating a biallelic pathogenic variant of SUMF1 gene is required for the diagnosis of MSD. Once a genetic mutation is confirmed in the affected child, parent carrier mutation testing is advised. If the parents wish to plan another biological offspring, the foetus may be tested for the same genetic variant by invasive prenatal testing, by chorionic villus biopsy. Earlier, in the absence of genetic confirmation, in lysosomal storage disorders, an enzyme assay on amniotic fluid would be performed [12].
We could not do enzyme analysis and exome analysis showed homozygous pathogenic variation in SUMF1 gene in our patient. Parents did not consent for carrier testing. Antenatal 2D and 3D ultrasonography, foetal 2D echocardiography may help identify associated findings such as certain cardiac defects or skeletal and facial dysmorphisms, which may later go on to require surgical intervention [13]. MSD is still an untreatable disease but multidisciplinary management is needed. Hence, identification, genetic confirmation and publishing newfound data is essential to aid better understanding of the clinical spectrum, the course, outcome, potential treatment approaches and also genetic counselling. Every suspected case of MSD must undergo a molecular genetic analysis of the SUMF1 gene. Understanding the genetic basis and molecular mechanism of MSD could help identify life-saving therapeutic approaches in the future.
This case highlights the importance of suspecting MSD in any neonate with skin changes in the form of ichthyosis, skeletal abnormalities, developmental delay and flat facies. Confirmation of diagnosis by enzyme estimation and/or genetic testing is essential to aid better understanding of the clinical spectrum, the course, outcome and counselling of the family. Prenatal diagnosis can be offered in future pregnancies if family mutation is known.
We have no conflict of interest to declare.
Copyright: © 2022 Srushti Gandh., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.