Ana Bernardo Ferreira1*, Teresa Amaral Pinheiro1, Ana Luísa Barbosa2, Catarina Matos de Figueiredo1, Sara Oliveira1 and Sónia Aires1
1 Pediatrics and Neonatology Department, Centro Hospitalar de Entre-o-Douro e Vouga, Portugal.
2 Internal Medicine Department, Centro Hospitalar de Entre-o-Douro e Vouga, Portugal.
*Corresponding Author: Ana Bernardo Ferreira, Pediatrics and Neonatology Department, Centro Hospitalar de Entre-o-Douro e Vouga, Portugal.
DOI: 10.31080/ASPE.2022.05.0503
Received: January 12, 2022; Published: February 25, 2022
Citation: Ana Bernardo Ferreira., et al. “A Transient form of Insulin-Requiring Diabetes Mellitus on a 15-Year-Old with Wolf-Hirschhorn Syndrome”. Acta Scientific Paediatrics 5.3 (2022): 03-06.
Wolf-Hirschhorn Syndrome is a rare disorder caused by a deletion in the short arm of chromosome 4. Depending on the genetic defect, phenotype variability can be extensive, with multi-systemic involvement. Diabetes mellitus classification depends on clinical characteristics, clinical course, and laboratory findings and has important implications. The authors report a case of a 15-year-old girl with Wolf-Hirschhorn Syndrome, who presented with Hyperglycemic Hyperosmolar Status and was diagnosed with diabetes. Insulin treatment was suspended four years after the diagnosis. The complexity of the case and the difficulties on classifying and understanding this transient form of pancreatic endocrine insufficiency are discussed.
Keywords: Wolf-Hirschhorn Syndrome; Diabetes Mellitus; Hyperglycemic Hyperosmolar Status; Diabetic Ketoacidosis; Transient Pancreatic Insufficiency.
WHS: Wolf-Hirschhorn Syndrome; DM: Diabetes Mellitus; T1DM: Type 1 Diabetes Mellitus of the Young; T2DM: Type 2 Diabetes Mellitus; HSS: Hyperglycemic Hyperosmolar Status; CDC: Centers for Disease Control and Prevention; UTI: Urinary Tract Infection; GAD: Glutamic Acid Decarboxylase; DKA: Diabetic Ketoacidosis; MODY: Maturity-Onset Diabetes.
WHS, originally described in the 1960s by Cooper and Hirschhorn [1], is a rare genetic condition caused by variable terminal deletions in the short arm of chromosome 4, covering the 4p16.3 region, known as the critical region (WHSCR). In most cases, it is due to a de novo deletion [2]. It is considered a contiguous gene syndrome, with correlation between deletion size and the spectrum and severity of the disease [3-5]. The loss of genetic material results in a series of clinical manifestations, some of them almost constant, others as part of a spectrum, variable among different individuals. Phenotype variability in this and others deletion syndromes, is still not totally understood. Some core characteristics are a variable degree of intellectual disabilities, seizures, growth deficiency and distinctive craniofacial features, giving the patients a greek warrior helmet appearance. Multiple other anomalies are part of the spectrum of the disease, namely congenital heart and genitourinary defects, skeletal abnormalities, among others [3,4].
DM etiology is heterogeneous and its classification depends on clinical features, laboratory findings and clinical course. Progressive pancreatic endocrine insufficiency and insulin resistance are the pathophysiological features of different types of DM. Most pediatric cases are T1DM, but T2DM is becoming increasingly important [6]. Other less common etiologies in children include monogenic diabetes syndromes, diseases of the exocrine pancreas, endocrinopathies, among others.
To our knowledge, there is no established link between WHS and any form of DM.
Our case is about a 15-year-old female with WHS. She is the third child of a non-consanguineous couple. As relevant family history, her mother was diagnosed with gestational diabetes during her pregnancy and later with T2DM and dyslipidemia, and her maternal grandmother had T2DM. She was born at 38-weeks gestation, weighting 2960g (10th-50th percentile) [Fenton curves]. At birth, a series of malformations was evident - wide bridge of the nose, short philtrum, micrognathia, low-set ears, cleft palate, microcephaly, sacrococcygeal dimple - and she had generalized hypotonia. Karyotype analysis revealed a terminal deletion of chromosome 4 (46, XX, del (4) (p14)), confirming WHS diagnosis. Her parents’ chromosomic study was normal, confirming a de novo deletion.
Global developmental delay was evident since birth. Despite early intervention, she failed to achieve developmental milestones. Expressive language was limited to guttural sounds and social interactions were poor. She never acquired the ability to walk. Feeding difficulties and poor weight gain presented soon. She remained below the 3rd percentile [CDC growth charts] for weight and length. At age thirteen she was diagnosed with dyslipidemia (total cholesterol 243 mg/dl and triglycerides 182 mg/dl) and started taking lovastatin, with near to normal lipid levels in subsequent studies.
At age fifteen, the patient was brought to the emergency department with drowsiness, food refusal, polydipsia and polyuria, since the day before. There was no history of weight loss, trauma, medication intake, fever, vomiting, diarrhea or steatorrhea. She was tachycardic, pale, obtunded and showed signs of mild dehydration. Blood pressure was normal and physical examination had no other relevant findings. Laboratory results are presented in table 1.
Serum lipase levels were not available. There was no ketonemia evaluation; urine dipstick revealed no ketonuria. Urinalysis showed glycosuria, leukocyturia and nitrituria; urine culture was positive for Escherichia coli. Chest radiograph had no relevant findings.
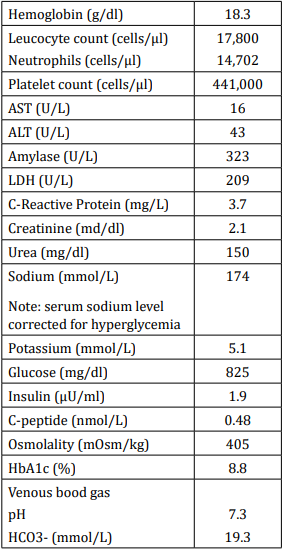
Table 1: Laboratory results at admission.
Note: Bold values are outside the normal range.
The proposed diagnosis was HSS as clinical presentation of DM, in association with UTI and complicated by acute renal lesion. Initial treatment included rehydration with a bolus of 20ml/kg of normal saline (0.9% NaCl), followed by administration of 0,45% saline for an estimated 10% fluid deficit plus daily maintenance fluid requirements. Total volume of fluid was calculated to be given over 48 hours. Insulin infusion started two hours after the start of fluid administration, initially at a rate of 0.1 units/kg per hour; 5% glucose was added to rehydration fluid and insulin dosage was titrated to maintain a gradual decrease in serum glucose concentration during the first hours of treatment. After 16 hours, glucose levels normalized and subcutaneous insulin (prandial and basal boluses) was initiated. She completed seven days of treatment with cefuroxime. Clinical status improved and oral intake was restarted two days later. Amylase levels normalized 24 hours after admission. Sodium levels and serum osmolality were slower in their decrease, taking about three days to stabilize in normal values. Acute renal lesion was rapidly resolved. She was discharged after 11 days with subcutaneous insulin according to a multiple daily injections regimen, with an insulin requirement of 0.5 units/kg. Insulin dosing was adjusted over the following weeks.
A month after hospital release, prandial boluses were not needed. The basal dose was progressively lowered, and about four years after DM diagnosis, insulin was suspended, with no record of subsequent hypo- or hyperglycemias. Initial HbA1c was 8.8%, and after one year, 7.1%. The following evaluations progressively decreased and the latest one, six months after stopping insulin administration, was 5.6%. Subsequent fasting C-peptide evaluations were lower (0,23 nmol/L) to normal (0,36 nmol/L), with concomitant glucose levels of approximately 100mg/dl. Pancreatic autoantibodies to insulin, islet cells and GAD were negative. Testing for celiac disease and autoimmune thyroiditis was negative. Next Generation Sequencing panel for Monogenic Diabetes studied 13 genes (ABCC8, BLK, CEL, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, KLF11, NEUROD1, PAX4, PDX1) and did not identify abnormalities.
We reported a complex case of a girl with WHS, who presented with an array of acute symptoms and laboratory findings that led to the uncommon diagnosis of HHS as presentation of DM, complicated by dehydration and acute renal lesion. A UTI was probably the triggering event, responsible for increasing metabolic stress. After four years of treatment, insulin was suspended, and glucose metabolism continued normal. The possibility of stopping insulin administration, years after the diagnosis, adds complexity to this case. Elevated HbA1c at presentation indicated a subacute or more prolonged hyperglycemic state, however the underlying cause for this transient glucose intolerance is still not defined.
The first diagnostic hypothesis was DKA. However, her extremely high serum glucose concentration and hyperosmolality without ketosis met the criteria for HHS [7]. HHS is a serious acute metabolic complication of decompensated DM, but it is considerably less frequent in children and adolescents than DKA [7]. Both share overlapping features, but it is important to differentiate them due to distinctive management strategies. Furthermore, HHS has a higher rate of complications and mortality [7,8]. Contrary to our case where a very short prodrome was present, symptoms of polyuria and polydipsia are typically more insidious in HSS, and the degree of dehydration may be underestimated, which can result in severe volume depletion. HHS is not a specific presentation of any type of diabetes. In cases of motor and intellectual disabilities, as in ours, recognizing the symptoms of HHS becomes a greater challenge, with potential for higher severity.
Although the genotype-phenotype relation in WHS is still not fully established, some studies attempted to attribute specific genetic defects to certain clinical aspects of WHS [9-11]. To the best of our knowledge, there is no definite genetic link between WHS and DM or pancreatic disease. However, mutations in chromosome region 4p16.1 are associated with DM, as seen in Wolfram Syndrome [12]. Genetic testing for MODY was negative. The question is whether or not the deletion of genetic material in our case, might be the cause or a predisposing factor for DM.
Personal history of dyslipidemia and family history of gestational diabetes and T2DM are features supporting the hypothesis of T2DM. However, there were no signs of insulin resistance and no history of obesity. Low levels of insulin and fasting C-peptide favored the possibility of T1DM, but the absence of pancreatic autoantibodies makes this diagnosis unlikely. To note that ZnT8, a more recent antibody in the investigation of DM, was not tested. In retrospect, type 3c diabetes due to pancreatitis was a postulated hypothesis.
We are aware of a case report of a girl with WHS and DM, who presented with HHS after severe acute pancreatitis [13]. In our case, elevated HbA1c indicated a subacute or chronic mechanism for DM, and chronic or acute recurrent pancreatitis were considered. Although there was no determination of liposoluble vitamins, there was no symptoms of malabsorption and no history of recurrent abdominal pain. Additionally, pancreatic endocrine insufficiency is an irreversible complication in the course of chronic pancreatic disease [14]. Hence, this diagnosis seemed less plausible.
WHS is a rare congenital disease, with an extensive array of genetic defects and phenotypic features. Classification of DM is of crucial importance for delineating long term management. We consider of great importance the discussion of different hypothesis for diabetes etiology and classification in this case due to its potential implications in delineating the relation between WHS and DM. Additionally, describing the complex presentation of DM in this case and discussing some features of HHS, might help to increase the index of suspicion and adequate management in similar cases.
The authors have no conflicts of interest to declare.
This work has not received any contribution, grant or scholarship.
Copyright: © 2022 Ana Bernardo Ferreira., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.