Piero Pavone1, Giuseppe Costanza2, Giovanni Cacciaguerra2*, Francesco Pizzo2, Maria Grazia Pappalardo2, Silvia Marino3, Rita Chiaramonte4 and Michele Vecchio4
1Department of Clinical and Experimental Medicine, University Hospital “Policlinico-San Marco”, Italy
2Postgraduate Training Program in Pediatrics, Department of Clinical and Experimental Medicine, University of Catania, Italy
3Unit of Pediatrics and Pediatric Emergency, AOU “Policlinico”, PO “San Marco”, University of Catania, Italy
4Department of Biomedical and Biotechnological Sciences, Section of Pharmacology, University of Catania, Italy
*Corresponding Author: Giovanni Cacciaguerra, Postgraduate Training Program in Pediatrics, Department of Clinical and Experimental Medicine, University of Catania, Italy.
Received: October 08, 2021; Published: November 15, 2021
Citation: Giovanni Cacciaguerra., et al. “Peripheral Neuropathies: State of the Art on Modern Use of Electrodiagnostic Examination in Guillain-Barrè Syndrome and Rehabilitative Implications in Children ”. Acta Scientific Paediatrics 4.12 (2021): 03-08.
Introduction: Guillain-Barré syndrome is an immune-mediated segmentary peripheral polyneuropathy The different variants depend on the physio-pathogenic involved mechanism (predominantly demyelinating, the type of nerve damaged (predominantly motor) the antibody associations (IgM and IgG GM1, GD1a, GD1b, GQ1b, GT1b, LM1) Electrodiagnostic examination (EDX) can distinguish the type of involvement of the peripheral nerves, and the secondary compromise of the muscles.
Objective: The purpose of this review is to analyze the literature of the last 10 years about the most current descriptive and diagnostic applications of EDX in children, useful also to project rehabilitative program and to monitor the prognosis in pediatric patients.
Keywords: Electrodiagnosis; Guillain-Barre Syndrome; Peripheral Nervous System Diseases; Polyradiculoneuropath
In childhood, an important category of peripheral nerve disorders is represented by the autoimmune neuropathies. The original classification of autoimmune neuropathies includes two main variants: the acute one of Guillain-Barré syndrome (GBS) and the chronic inflammatory demyelinating polyradiculoneuropathy (CIPD). Other variants are classified according to the different clinical, laboratory (autoantibodies) and electrophysiological characteristics [1].
GBS is an immune-mediated segmentary peripheral polyneuropathy, with an incidence of 0.34 - 1.34 cases/100.000 in children [2].
In about two thirds of cases, a cross-reaction mechanism related to a previous infection of Campylobacter jenuni, Cytomegalovirus, Epstein-Barr virus, or other bacteria (Haemophilus influenzae, Mycoplasma pneumoniae), within 3 weeks from onset, could cause an autoimmune reaction. The cerebrospinal fluid is characterized by the albumino-cytologic dissociation, an elevation of the cephalorachidian protein with contextual normal white blood cell count, usually not present at the disease onset [3].
The typical form is the acute inflammatory demyelinating polyradiculoneuropathy (AIDP), which represents roughly 85% of cases in the western world in pediatric population. The symptomatology is characterized by an acute ascending weakness at onset, sometimes a progressive loss of sensation and areflexia, within a couple of months [4].
The different variants depend on the physio-pathogenic involved mechanism (predominantly demyelinating), the type of nerve damaged (predominantly motor), antibody associations (IgM and IgG GM1, GD1a, GD1b, GQ1b, GT1b, LM1) and the specific symptomatology.
The most frequent sub-variants are Miller-Fisher syndrome (MFS) and axonal motor acute neuropathy (AMAN). MFS is characterized by the characteristic triad: ophthalmoplegia, areflexia and ataxia, in absence of weakness [4]. The pathogenesis of MFS is related to the presence of anti-GQ1b antibodies in up to 85% of cases [5]. MFS has clinical and laboratory aspects in common with Bickerstaff brainstem encephalitis, which shows also central nervous system involvement [6].
In the AMAN, there is an exclusive impairment of motor axons, without sensitivity disorders. It is characterized by a good clinical course and a favorable prognosis. There is a strong association with a previous Campylobacter jejuni infection and the presence of GD1a, GM1 or GD3 gangliosides autoantibodies, presented in about 50% of cases [7].
Rare forms of GBS include the axonal motor-sensitive acute neuropathies (AMSAN), associated to anti-GD1a and anti-GM1 antibodies, and the autonomic neuropathies associated to nicotinic acetylcholine receptor antibodies [1]. The main variants with the respective specific autoantibodies are summarized in table 1.
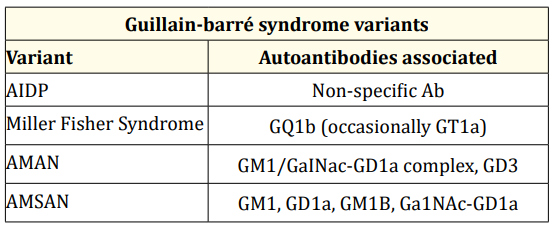
Table 1: Guillain-Barrè Syndrome Variants.
AIDP: Acute Inflammatory Demyelinating
Polyradiculoneuropathy; AMAN: Axonal Motor Acute Neuropathy;
AMSAN: Axonal Motor-Sensitive Acute Neuropathy.
EDX can help physicians to a obtain a specific diagnosis,and followed treatment. EDX permits to distinguish the type (axonal or demyelinating) and the localization of the peripheral nerves’ involvement [8].
EDX consists of two main parts: nerve conduction studies (NCS) and needle electromyography.
In NCS, the peripheral nerve is stimulated with a transcutaneous electrode for very short duration. It analyzes the following parameters.
Compound muscle action potential (CMAP) is also known as an M-response. In 1971, McComas., et al. [9] described an attractively simple method for counting motor units. The authors showed that was possible to arrange was possible to arrange for the recorded electrical activity derived from a single muscle and for the evoked potentials and then to summate algebraically [9]. They measured the amplitudes of the muscle action potentials generated, firstly, by a single motor unit and, secondly, by the whole muscle, then the number of motor units within the muscle can be determined by a division [9]. The amplitude of the resultant response was divided by the number of increments to yield an estimate of the amplitude of a single unit; this value was divided by the maximum CMAP to estimate of the number of motor units [9]. More recently a new examination system was applied in the clinical practice: the incremental motor unit number estimation (MUNE) technique was first applied to upper extremity muscles supplied by median, ulnar and radial nerves. MUNE techniques have been used to quantify the proportion of surviving lower motor neurons. The results of multiple studies have confirmed that MUNE, when applied longitudinally, may reflect the rate of disease progression [10].
In the needle electromyography, a concentric needle electrode is inserted into the target muscle and its electrical activity is recorded during rest and periods of maximal and minimal voluntary muscle contraction. Needle electromyography is commonly used to assess axonal nerve lesions [16].
Parameters analyzed are:
According to the electrodiagnostic criteria established by Hadden., et al. [17], GBS can be classified in primary demyelinating 05 GBS, AIDP, GBS with primary axonal damage, and AMAN (Table 2).

Table 2: Hadden criteria.
DML: Distal Motor Latency; NCV: Nerve Conduction Velocity;
CMAP: Compound Muscle Action Potential; LLN: Lower Limit Of
Normal; ULN: Upper Limit of Normal.
However, these criteria have been further developed by Rajabally., et al. [4]. who have shown comparable rates of acute inflammatory AIDP, axonal GBS and other different forms between single nerve conduction studies compared with electrophysiologic diagnosis. The classification with the modified criteria proposed by Rajabally., et al. [4] demonstrated that single electrophysiological studies could be sufficient to establish the definitive diagnosis of the different GBS subtypes.
The purpose of this narrative review is to analyze literature of the last 10 years and we about the most current descriptive and diagnostic applications of EDX in children, useful also to project the rehabilitative program and monitor the prognosis in pediatric patients.
A literature review was performed on three the following medical electronic databases PubMed, Cochrane Library, Scopus, and Web of Science by three authors (G.C, G.C, F.P), evaluating the studies published in the period between January 2010 and January 2020. In the search bar, we first introduced the term “electrodiagnosis” OR “electrodiagnostic” OR “nerve conduction” OR “electromyography” AND “neuropathies” OR “Guillain-Barrè” AND “children”, and a number of 30 elegibles manuscripts were collected.
Qualified studies for the present review were performed searching in databases selecting with a screening of the titles and abstracts through the following inclusion criteria: publications within the last 10 years, written in English language, studies published in peer-reviewed journals reporting clinical or pre-clinical outcomes. Exclusion criteria were articles without EDX as a primary outcome or systematic review involving a similar subject. In addition, we ignored studies with no obtainable data or without an accessible full text. We also excluded all the remaining duplicates.
The study selection and the data extraction were performed independently by two authors (G.C.; A.S.), and any conflicts were resolved by discussion amongst the authors. The senior investigators (P.P., M.V., R.C.) were consulted to re-examine the full process. We followed the rules of the Declaration of Helsinki and the Prisma flow diagram of the literature selection and review process checklist from Moher, D., et al. (2009) [Figure 1].

Figure 1: Flow diagram of literature selection and review process.
The review described the diagnostic applications of EDX in children with GBS, useful to propose the rehabilitative program, and monitor the prognosis in pediatric patients.
Ashrafi., et al. [14] used electrophysiological analysis, to classify GBS in primary demyelinating, AIDP, GBS with primary axonal damage, and AMAN). based on Hadden’s research [17].
Most AIDP patients exhibited the following electrophysiological characteristics: a pattern of prolonged F wave latency with low persistency, absence of SNAP, reduced CMAP, prolonged DML, reduced NCV, and abnormal temporal dispersion or conduction block.
On the other hand, most AMAN patients showed reduced CMAP amplitude, F wave with normal latency and reduced persistency, normal SNAP amplitude, normal distal latency, normal sensory NCV, and conduction block or temporal dispersion [17].
Roodbol., et al. [18]. used NCS to evaluate 67 children in a retrospective cohort to describe the diagnostic features of pediatric GBS and validate the Brighton criteria [19]. The NCS showed the classical signs of GBS: a polyradiculoneuropathy in 48 patients, The NCS were normal in five cases. The predominant subtype was AIDP, present in 53 patients, followed by AMAN in 2 children. One child showed AMSAN-features. In 12 children the NCS was unclassifiable. In 2 children with persistent paraparesis, NCS indicated a demyelinating polyneuropathy of both arms and legs [18].
Karalok., et al. [20] used EMG to identify the subtype of GBS. Motor nerve conduction (MNC) studies were performed on the median, ulnar, posterior tibialis, and common peroneal nerves. On the posterior tibial nerves were studies H responses. Sensory nerve conduction (SNC) was analyzed in the median, ulnar, and sural nerves. F-wave response studies were performed on the tibial nerves [20]. All parameters were compared with age-related normal values.
In children of 1-2 years, nerve conduction parameters only approach 80-90% of the adult values, so normal limits were set at 80% of the values for NCV (CMAP amplitudes, and 120% of the values for distal latencies). For children younger than 1-year old, normal limits were set at half the values for NCV and CMAP amplitudes, and twice the values for distal latencies. Needle electromyography (EMG) was performed in all patients for at least two proximal and two distal limb muscles [20].
Karalok., et al. [20] evaluated 54 patients, classified into five groups:
Estrade., et al. [21] performed a multicenter study at the Montpellier and Toulouse University Hospitals. They conducted a retrospective investigation on one hundred-ten children with GBS syndrome by collecting medical records from the period between January 2000 and June, 2016. Their study aimed to describe the clinical characteristics and the long-term sequelae of GBS in a French pediatric population. EDX identified 70% of children affected AIDP and 16% by AMAN. Moreover, this study [21] showed that, although AIDP and AMAN have similar clinical manifestations, the axonal form (AMAN or AMSAN) was associated with a higher risk of long-term sequelae [21].
Kılıç., et al. [22] showed etiology, clinical and electrophysiological findings after treatment and prognosis of the patients with GBS. This retrospective study included 20 female and 25 male patients, with GBS. In pediatric patients, an important and independent risk factor for mechanical ventilation is the autonomic dysfunction. In their study, six (13.3%) patients required ventilation support. Autonomic dysfunction was seen in all these patients, and bilateral facial paralysis was present in four patients. In their patients, AMAN subtype was characterized by cranial nerve involvement and those patients had mechanical ventilation requirements. Cranial nerve involvement was also found in the AIDP variant but less important than in AMAN patients [22].
Barzegar., et al. [23] described the predictors of disability, such as the recovery of independent walking, autonomic and cranial nerve involvement, electromyographic findings. at 2 and 6 months after the onset of GBS. This investigation showed that 90.5% of patients could walk independently at 6 months and the mean duration of independent walking was 2.97 ± 3.02. In the univariate analysis, poor walking outcome at 6 months was associated with a disability score of 43 (P = 0.03), autonomic nerve involvement (P = 0.003), cranial nerve involvement (P = 0.008), and absent CMAP (P = 0.048). The multivariate analysis highlighted that low functional outcome was independently associated with cranial nerve involvement (P = 0.008) and absence of CMAP (P = 0.022) [23].
The electrodiagnostic studies are always more important, thanks to a better knowledge of the pathology. Electromyography helps to make a correct and rapid classification, diagnosis, risk stratification of ascending and worsening evolution and to propose a specific rehabilitation program process tailored to the patient’s needs. Other studies on the application of EDX are needed to propose diagnostic and therapeutic protocols to guide physicians to better follow children with GBS.
Conceptualization, G.C., F.P., and P.P.; methodology, G.C.; investigation, G.C.; data curation, M.V.; writing—original draft preparation, P.P., G.C., M.V.; writing—review and editing, F.P., G.C., G.C.; supervision, P.P., M.V., RC. All authors read and approved the final submitted version.
This research received no external funding.
The authors would like to thank AME (USA) Editor American manuscript Editors, for editing the manuscript.
The authors declare no conflicts of interest.
Copyright: © 2021 Giovanni Cacciaguerra., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.