Amal Badre*, T Faid, M Lehlimi, M Chemsi, A Habzi and S Benomar
Neonatal Medicine and Resuscitation Department, Abderrahim Harouchi Children's Hospital, Ibn Rochd University Hospital, Casablanca, Morocco
*Corresponding Author: Amal Badre, Neonatal Medicine and Resuscitation Department, Abderrahim Harouchi Children's Hospital, Ibn Rochd University Hospital, Hassan II University Casablanca, Casablanca, Morocco.
Received: June 03, 2021; Published: August 02, 2021
Citation: Amal Badre., et al. “Aplasia Cutis Congenita of the Scalp Revealing Trisomy 13”. Acta Scientific Paediatrics 4.9 (2021): 47-50.
Aplasia cutis congenita (ACC) is an uncommon congenital malformation affecting mainly the scalp in front of the lambda fontanelle. It is characterized by a focal absence of epidermis, dermis, and in some cases subcutaneous tissues, including bone and dura mater. This malformation is unique in 75%, but it may be part of a poly malformation syndrome. Aplasia cutis congenita represents a clinical sign that may be indicative of bone agenesis or severe genetic abnormalities. We report in this article the case of a male newborn, issued from a non-consanguineous marriage, a non-monitored pregnancy, without any drug intake during pregnancy, vaginal delivery with a birth weight of 2500g. Our patient presents with occipital aplasia cutis congenita, associated with facial dysmorphia, microphthalmia, hypotonia, polydactyly and agenesis of the left kidney, his karyotype revealed the presence of trisomy 13.
The aim of this article is to study the clinical, therapeutic and progressive aspect of this rare congenital disorder.
Keywords: Aplasia; Cutis Congenital; Trisomy 13
Aplasia cutis congenita (ACC) is a rare congenital malformation represented as a deficiency that may affect different layers of cutaneous tissue. This malformation affects most commonly the scalp (mainly in front of the lambda fontanelle) but may more exceptionally occur in different body parts. It is characterized by localized and well-demarcated loss of substance of varying severity, ranging from an absence of epidermis to the involvement of deeper elements such as bone and dura mater [1]. The diagnosis of this malformation is essentially clinical, and a biopsy is usually not needed. This defect occurs in isolation in 75% of cases, but it may in some cases be part of a heterogeneous group of syndromes and may provide a clue to an underlying disorder.
We aim in this report to study the clinical, therapeutic and evolutionary characteristics of this rare congenital disorder.
We report the case of a male premature infant born via vaginal delivery at 35 weeks of gestation to a 28-years-old mother after a non-monitored first pregnancy. Parents are unrelated, and no record of maternal morbidities or intake of suspected substances during the pregnancy were reported. The initial examination showed a premature baby, with a birth weight of 2500 g. Right after his birth, the newborn suffered a respiratory distress with a Silverman scale at 3/10. Dermatological examination revealed an ulcerated, erythematous, non-hemorrhagic skin defect, measuring 3.0 × 2.3 cm over his scalp vertex (Figure 1), associated with an unusual head shape, microphthalmia, hypotonia, and polydactyly. The abdominal ultrasound had shown an agenesis of the left kidney, and the karyotype revealed the presence of trisomy 13. The lesion over his scalp was conservatively treated with normal saline cleansing and covered with gauze during hospitalization. The echocardiogram was not realised and the evolution of the lesion was not precise since the patient died at the first week of life because of neonatal sepsis.
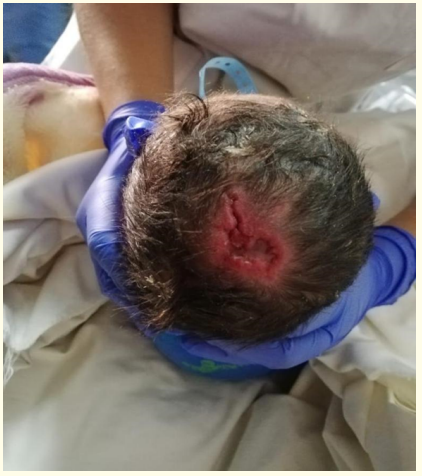
Figure 1: A 3.0 × 2.3 cm skin defect on scalp vertex.
Aplasia cutis congenita is a rare birth defect, with an incidence estimated at 1/10 000 births and a prevalence of 1 case per 5000 [2]. Described as the focal absence of skin noticed at birth, it was reported for the first time by Cordon with the involvement of limbs in 1767 [3], in 1826 Campbell first described the aplasia cutis congenita of the scalp [4], which is reported as the most common site for this defect in nearly 90% of the cases [5].
The exact mechanism of this defect is still not completely understood. An etiological classification based on the location of skin defect and the presence or absence of other malformations proposed by Frieden divides this pathology into nine groups [5].
The lesion in the aplasia cutis congenita is mainly found to be isolated, however it can be a part of various malformation syndrome and reveals a specific etiology [8]. These etiologies may be genetic where the aplasia cutis congenita take part of a poly malformation syndrome in the context of a chromosomal abnormality, as it is shown in many cases of genetic conditions, for instance the Adams-Oliver syndrome which associates the aplasia cutis congenita to terminal limb defects [6] and Trisomy 13 also called Patau syndrome characterized by the presence of one extra chromosome 13 in group D chromosome with the association of the aplasia cutis congenita to many other possible clinical malformations: severe defect in the central nervous system development, holoprosencephaly, low birth weight, severe mental retardation, microcephaly, microphthalmos or anophthalmos, cleft lip and palate, cardiovascular malformation and polycystic kidney. This syndrome is frequently fatal, as 50% of affected children die within one month after their birth and 90% die within one year [7].
Otherwise, the cause of this defect may be non-genetic, including traumatic mechanism, intrauterine infections with varicella zoster or herpes viruses, amniotic defects, thrombotic events, and teratogen substances used during pregnancy [4-6]. In 1986, Frieden proposed a classification system for ACC that included nine groups based on the lesion characteristics, associated abnormalities, and type of inheritance (Table 1) [9,10].
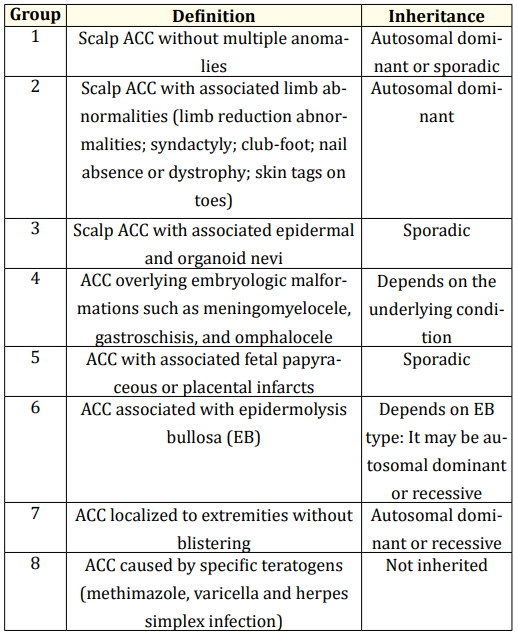
Table 1: Classification of aplasia cutis congenita (Adapted from Frieden’s Classification).
Diagnosed at birth, in the Lalla Meryem maternity unit of the A. Harrouchi Mother and Children’s Hospital in Casablanca Morocco, the aplasia cutis congenita was associated to other malformations represented by an unusual head shape, a facial dysmorphia, a polydactyly, a microphthalmia, a hypotonia, and a polydactyly, also an agenesis of the left kidney. In front of this poly malformation syndrome a chromosomal anomaly as an etiology of aplasia cutis congenita is suspected and confirmed the diagnosis of trisomy 13 by the karyotype that reveals the presence of an extra chromosome 13 in the group D. the patient under analysis in this study seems to fit in group 9 of Frieden’s classification.
The diagnosis of this pathology is mainly based on physical examination at birth in general, with the discovery of an isolated loss of skin substance or an isolated thin membrane disrupting the skin regularity of the scalp. The border with normal skin is abrupt and the skin tissue is reduced to its simplest form. In-depth, the lesion varies from a simple absence of the skin to the involvement of dura mater in severe forms. Associated examinations such as transfontanellar ultrasound and head-CT may be performed to seek for neurological defect or anatomic abnormalities [11].
Acute complications are mainly represented by local infection or bleeding, and the most frequent late complications are alopecia and hypertrophic scars usually non reversible, but more severe complications that can be fatal may be observed in some cases, for instance, infections of the central nervous system, meningitis, thrombosis, or haemorrhages of the sagittal sinus [12].
The prognosis of this defect depends on the extension and the depth of the lesion, associated malformations, and the quality of the management. The mortality rate is reported at 20 - 50% [6]. Superficial lesions limited to the epidermis may heal spontaneously with local antibiotic therapy and dressing. In the skin areas exceeding 4cm², an excision followed by a thin graft may be necessary to accelerate healing and reduce hospitalization time. An early surgical attitude associating a debridement of necrotic areas, and a covering with local flaps of the scalp or graft thin skin may be adopted in the case of absent bony crust with possible exposure of the underlying structures, with the life-threatening risk of ulceration of longitudinal sinus [13].
Aplasia cutis congenita is a physical finding, it is in general an isolated skin defect but it can be part of a poly malformation syndrome and may have many etiologies, despite its exceptional character, this neonatal defect must be known and carefully taken care of, as it can be life-threatening to the newborn.
Copyright: © 2021 Amal Badre., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.