Masayo Yamazaki1*, Makiko Oguma1, Koji Yokoyama1, Natsuko Ichinoi2, Atsuo Kikuchi2, Shigeo Kure2, Toshihiro Tajima1 and Takanori Yamagata1
1Department of Pediatrics, Jichi Medical University, Shimotsuke, Tochigi, Japan
2Department of Pediatrics, Tohoku University Graduate School of Medicine, Aoba, Sendai, Miyagi, Japan
*Corresponding Author: Masayo Yamazaki, Department of Pediatrics, Jichi Medical University, Shimotsuke, Tochigi, Japan.
Received: June 21, 2021; Published: July 08, 2021
Citation: Masayo Yamazaki., et al. “Growth Hormone Deficiency and Citrin Deficiency: A Case Report”. Acta Scientific Paediatrics 4.8 (2021): 24-28.
Citrin deficiency (CD) is an autosomal recessive disorder that causes a variety of symptoms such as neonatal intrahepatic cholestasis caused by CD (NICCD), failure to thrive, hypoglycemia, and growth failure. We report on a young female with CD complicated with growth hormone deficiency (GHD).
The patient developed afebrile seizure due to hypoglycemia with short stature at three years of age. Detailed medical examinations were carried out. Amino acid levels were within normal range. Biochemical evaluation showed GHD and central hypothyroidism. In the neonatal period, icterus was not indicated. The mother was interviewed; there seemed to have been repeated hypoglycemic episodes. Thereafter, GH and levothyroxine supplementation were initiated. Hypoglycemia was not improved. The patient’s preference for a high-protein, high-fat, and low-carbohydrate diet was noticed during follow-up. CD was suspected, and genetic analysis confirmed the diagnosis of CD.
As hypoglycemia and growth failure are both indicative of CD, it is necessary to consider CD when looking at both of them. In addition, GHD may be involved in growth failure of a patient’s CD. Regarding of this point, further studies of patients with CD are required.
Keywords: Citrin Deficiency; Hypoglycemia; Short Stature; Growth Hormone Deficiency; Mitochondrial Dysfunction
CD: Citrin Deficiency; NICCD: Neonatal Intrahepatic Cholestasis; GHD: Growth Hormone Deficiency, CTLN2: Adult-Onset Type II Citrullinemia; FTTDCD: Failure to Thrive and Dyslipidemia Caused by Citrin Deficiency; AGC2: Aspartate-Glutamate Carrier 2; ATP: Adenosine Triphosphate; NADH: Reduced Nicotinamide Adenine Dinucleotide; SD: Standard Deviation; PKU: Phenylketonuria; MSUD: Maple Syrup Urine Disease; ER: Emergency Room; MRI: Magnetic Resonance Imaging; BE: Base Excess; IGF-1: InsulinLike Growth Factor 1; ACTH: Adrenocorticotropic Hormone; TSH: Thyroid Stimulating Hormone; CRH: Corticotropin-Releasing Hormone; TRH: Thyrotropin-Releasing Hormone; PRL: Prolactin; DNA: Deoxyribonucleic Acid; MCT: Medium Chain Triglyceride; MELAS: Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes; KSS: Kearns-Sayre Syndrome; mRNA: Messenger Ribonucleic Acid
Citrin deficiency (CD) is an autosomal recessive disorder caused by a pathogenic variant of SLC25A13 [1]. In age-dependent CD there are two clinical phenotypes: neonatal intrahepatic cholestasis caused by CD (NICCD: OMIM 605814) and adult-onset type II citrullinemia (CTLN2: OMIM 603417) [2]. About 50% of children with CD develop NICCD from the neonatal period until around six months of age. While most of patients become asymptomatic afterwards, some patients have recurrent episodes of hypoglycemia, failure to thrive, and dyslipidemia (FTTDCD) [3]. The remaining patients with CD but without NICCD are often diagnosed by characteristic food preferences or recurrent hypoglycemia [4].
Citrin is a mitochondrial aspartate-glutamate carrier 2 (AGC2) on the inner mitochondrial membrane. AGC2 is an important component of the malate-aspartate shuttle. The shuttle transfers reducing equivalents of reduced nicotinamide adenine dinucleotide (NADH) from the cytosol to the mitochondrial matrix as the “NADH shuttle” [5]. AGC2 deficiency impairs glycolysis and de novo lipogenesis. Numakura., et al. [6] reported that the impairment of de novo lipogenesis is the main cause of growth failure in children with CD.
Here we report on a Japanese girl with CD complicated with growth hormone deficiency (GHD) and central hypopituitarism. To our knowledge, there is no previous report of a case of CD combined with GHD.
The patient had no family history of CD or endocrine diseases. She was delivered at 36 weeks of gestation by Caesarean delivery because of a nonreactive nonstress test, her breech presentation, and her mother’s schistorrhachis. The Apgar scores at one minute, five minutes, and 10 minutes were 8, 8, and 9, respectively. Her birth weight was 2358g (-0.0 standard deviations (SD) for a normal Japanese girl of the gestational age) and her birth length was 42.0 cm (-1.7 SD for a normal Japanese girl of the gestational age). She was not dysmorphic. At four days of age she was placed under phototherapy for one day, to treat icterus. She had no episodes of hypoglycemia. She was discharged from the hospital at five days of age. The neonatal blood screening for phenylketonuria (PKU), maple syrup urine disease (MSUD), homocystinuria and galactosemia [7] was normal.
At one month of age she was brought to our hospital because of poor weight gain. Her mother complained that formula feeding put her daughter in a bad mood. Thereafter she had regular follow-ups to check her weight and amount of milk.
At one year and six months old she was taken by her mother to the emergency room (ER) because of loss of consciousness. She had looked pale and exhibited a poor response for a while. As she was alert during examination at the ER, she was not subjected to further medical examination. At one year and seven months of age she was seen again in the hospital because of her short stature. (Her height was 71.0 cm, -3.3 SD for a normal Japanese girl of this age. Her weight was 8.3 kg, -1.7 SD for a normal Japanese girl of this age) (See figure 1). Magnetic resonance imaging (MRI) of the brain, including the pituitary gland, gave normal results. Biochemical examination, including blood sugar and liver function, showed that all was normal at this time. At three years of age the subject was urgently taken to hospital after an afebrile seizure one morning, before breakfast. The initial blood analysis showed her blood glucose level was 1.1 mmol/L with insulin < 3.47 pmol/L. Her blood gas analysis showed: pH 7.242; pCO2 37.2 mmHg; HCO3- 15.7 mmol/L; base excess (BE) -10.9; and NH3 44 μmol/L, which indicated metabolic acidosis. Her dipstick urinalysis detected ketones at a scale of 2+, which indicated ketotic hypoglycemia. After bolus administration of intravenous glucose, she recovered from the seizure. Further investigation for ketotic hypoglycemia was carried out. The endocrinological evaluation is summarized in table 1. Amino acid levels and urine organic acid analysis were not specific. Because she had hypoglycemia, slightly low IGF-1 and short stature, she was suspected to have a combined pituitary hormone deficiency. Stimulation tests were carried out with arginine, corticotropin-releasing hormone (CRH), and thyrotropin-releasing hormone (TRH). As shown in table 1, she had GHD. In addition, the TSH response to TRH showed prolonged secretion, indicative of central hypopituitarism (Table 1). GH and levothyroxine supplementation were started (Figure 1).
Although her height velocity improved slightly, as shown in figure 1, episodes of hypoglycemia even under GH treatment occurred often, and the patient and her mother complained that she became fatigued easily, compared with other children. Uncooked cornstarch was added to her diet, but the frequency of hypoglycemia did not change. Because GH treatment did not resolve hypoglycemia, congenital metabolic diseases were suspected. At seven years of age, the patient’s amino acid, urine organic acid and acylcarnitine profile were analyzed. The results of these examinations were non-specific.
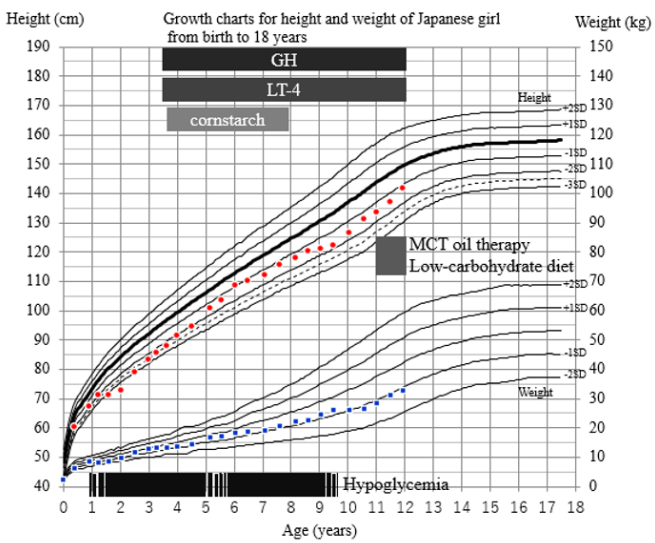
Figure 1: Growth charts, the course of treatment, and symptoms.
The patient started to have recurrent episodes of hypoglycemia from around one year of age. At three years of age she was taken to our ER
because of seizure due to hypoglycemia. Because she was also of short stature we conducted a pituitary stimulation test, which showed
GHD and central hypothyroidism. GH and levothyroxine supplementation began but she continued to have hypoglycemia. We added a
supplement of uncooked cornstarch, which was not effective. From eight years of age, she rarely showed hypoglycemia. When the patient
was 11 years old we made a diagnosis of citrin deficiency by genetic analysis. MCT oil supplementation began.

Table 1: Endocrinological findings in the patient.
The results of the endocrinological analysis and pituitary stimulation test with arginine, CRH, and TRH. PRL: prolactin.
After the age of eight years, episodes of hypoglycemia ceased; it seemed to have resolved spontaneously. A careful review of the patient’s diet revealed that she did not like rice or sweet snacks, and preferred a high-protein, high-fat diet, especially peanut. Based on these eating preferences, and on hypoglycemia and her short stature, CD was suspected. After receiving approval from the institutional review board and obtaining written informed consent, deoxyribonucleic acid (DNA) diagnosis for known SLC25A13 (NM_014251.3) [8] was performed when she was 11 years old. She was a compound heterozygote for c.1177+1G>A, p.(340_392del) and c.1230+1G>A, p.(411_437del), confirming the diagnosis of CD. Medium chain triglyceride (MCT) oil supplementation was started (Figure 1). After MCT oil supplementation, she became fatigued less easily and her daily activity increased. GH treatment is being continued.
This is the first reported case of CD complicated with GHD. A peculiar eating habits is a characteristic symptom of an apparently healthy period in CD patients [6]. In addition, ketotic hypoglycemia is thought to be a common symptom [4,9]. Our patient showed ketotic hypoglycemia and seizure at three years of age, but before that she had episodes of pallor and drowsiness, suggestive of hypoglycemia. She also presented with growth failure. One of the differential diagnoses of ketotic hypoglycemia was GHD. At the first medical examination, secretion of GH was impaired, leading to the diagnosis of GHD. GH therapy was likely to be partially effective for growth, but hypoglycemia and easy fatigue were not resolved. In the subsequent course, CD was suspected because of the patient’s peculiar eating habits, and the diagnosis of CD was confirmed by the genetic analysis of SLC25A13. There are reports of some patients being initially diagnosed with ketotic hypoglycemia and then finally diagnosed as having CD [9]. The diagnostic clue in these cases was a specific eating habits similar to our case. However, since peculiar eating habits may not be evident in early childhood, CD should always be considered in the differential diagnosis of ketotic hypoglycemia.
Our patient had GHD and central hypothyroidism, suggestive of endocrinological dysfunction in the hypothalamic-pituitary region. Numakura., et al. [6] reported that patients with CD have short stature. According to their study, patients unaffected by NICCD showed low birth weight and height and growth impairment. In particular, the height of girls unaffected by NICCD was significantly low, from two months to 12 years of age. These findings are in agreement with our case. The impairment of growth was due to a defect in glycolysis and de novo lipolysis, leading to an energy deficit. A literature search revealed no reports that evaluated GH secretion in CD patients. Citrin, encoded by SLC25A13 [1], is a mitochondrial aspartate-glutamate carrier 2 (AGC2) on the inner mitochondrial membrane. AGC2 is an important component of the malate-aspartate shuttle. The shuttle transfers reducing equivalents of NADH from the cytosol to the mitochondrial matrix as the “NADH shuttle” [5]. NADH transported into the mitochondria is used in the electron transport chain to produce adenosine triphosphate (ATP). Therefore, CD leads to the dysfunction of the NADH shuttle, which results in reduced ATP production, which can lead to mitochondrial dysfunction. Patients with mitochondrial diseases, such as mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (together, MELAS), and Kearns-Sayre syndrome (KSS), are known to have hypothalamus-pituitary dysfunction [10]. Indeed, some patients with mitochondrial disease have been reported to have GHD. While messenger ribonucleic acid (mRNA) of SLC25A13 is highly expressed in the liver, it is also expressed in the brain, including the hypothalamus and pituitary [11]. In addition, AGC2 expression was observed in the hypothalamus of mice [12]. Based on these findings, citrin deficiency may cause hypothalamic dysfunction, leading to GHD and central hypothyroidism via mitochondrial dysfunction. However, the possibility that CD and GHD occurred together by chance cannot be excluded. After the achievement of adult height, endocrinological reevaluation will be necessary.
GH therapy seems to be partially effective for catch-up growth, but not for hypoglycemia. As reported, since MCT oil supplementation is effective for energy deficit, it may be the first choice for catch-up growth. However, since CD patients show severe short stature, it should be carefully considered whether or not to evaluate GH secretion.
In conclusion, as hypoglycemia and growth failure are signs of CD, it is necessary to consider CD when patients show both. In addition, GHD may be involved in growth failure in patients with CD. With regard to this point, further studies in CD patients are required.
The authors declare that they have no competing interests.
Copyright: © 2021 Masayo Yamazaki., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.