Gayatri Nerakh1*, Santosh Rajeev P2, Sowjanya SVNS2, and Tejopratap Oleti2
1Consultant Geneticist, Department of Fetal Medicine and Genetics, Fernandez Hospital, Hyderabad, India
2Consultant Neonatologist, Department of Neonatology, Fernandez Hospital, Hyderabad, India
*Corresponding Author: Gayatri Nerakh, Consultant Geneticist, Department of Fetal Medicine and Genetics, Fernandez Hospital, Hyderabad, India.
Received: April 24, 2021; Published: June 23, 2021
Citation: Gayatri Nerakh., et al. “A Rare Mitochondrial Disorder in a Newborn with Facial Dysmorphism and Persistent Respiratory Distress”. Acta Scientific Paediatrics 4.7 (2021): 41-43.
Mitochondrial progressive myopathy with congenital cataract, hearing loss and developmental delay (MPMCD) is a rare autosomal recessive mitochondrial disorder. There are only few reports of GFER associated MPMCD. We report a neonate born second in order to seventh degree consanguineous couple who had severe persistent respiratory distress, metabolic acidosis and succumbed on day 5. The neonate had flat face, congenital cataract with narrow chest, brachydactyly of fingers and toes and bilateral varus deformity. Neurological examination revealed hypotonia with diminished reflexes. Arterial blood gas analysis showed metabolic acidosis. There was elevated lactate in plasma and in urine GC-MS. Clinical exome sequencing revealed a likely pathogenic homozygous variant [NM_005262] c.580C>T (p.Arg194Cys) in exon 3 of GFER gene. High index of suspicion is crucial for recognition of this disorder as identification of the exact underlying etiology aids in appropriate genetic counseling of the couple.
Keywords: Flat Face; Congenital Cataract; MPMCD; GFER Gene
MPMCD: Mitochondrial Progressive Myopathy with Congenital Cataract, Hearing Loss and Developmental Delay; GC-MS: Gas Chromatography-Mass Spectrometry; DNA: Deoxyribonucleic Acid; mtDNA: Mitochondrial DNA; NT: Nuchal Translucency; PAPPA: Pregnancy-Associated Plasma Protein-A; mmol/l: Millimoles/Litre; CADD: Combined Annotation Dependent Depletion
Mitochondrial disorders are a clinically heterogeneous group of disorders caused by mutation of genes encoded by either nuclear DNA or mitochondrial DNA. Most of the mitochondrial disorders involve multiple organ systems and often present with neurological and myopathic features [1]. GFER codes for mitochondrial disulfide relay system protein. Mutations in GFER causes reduced mitochondrial complex I, II, and IV activity specifically in reducing complex IV activity in humans, abnormal ultrastructural morphology of the mitochondria, mitochondrial instability and accelerated accumulation of multiple mtDNA deletions resulting in Mitochondrial progressive myopathy with congenital cataract, hearing loss and developmental delay, an infantile mitochondrial disorder [2]. It is a rare autosomal recessive genetic disorder characterized by facial dysmorphism, congenital cataract, progressive muscular hypotonia, persistent lactic acidosis. Neonates who survive have global development delay and sensorineural hearing loss.
This is a case of neonate second born to healthy seventh degree consanguineous couple. The first child of the couple is healthy. The family history was unremarkable. During antenatal period NT scan, anomaly scan and growth scans were normal. Combined first trimester serum screen was normal except for low PAPPA levels. The neonate was born by preterm vaginal delivery at 34 weeks with the birth weight of 1.6 kg corresponding to 9th centile. The neonate had severe respiratory distress, was shifted to neonatal intensive care unit and kept on oxygen initially. In view of high oxygen dependency, the neonate was ventilated. There was no jaundice or seizures. On examination of the neonate, there was flat face with broad prominent forehead, posteriorly rotated ears, telecanthus with slight upslant of eyes with corneal clouding and bilateral cataract, depressed flat nasal bridge and flat philtrum (Figure 1A-1C). Chest was narrow. Neck and spine were normal. There was brachydactyly with tapering of fingers, bilateral varus deformity of foot with brachydactyly of toes and mild joint laxity. On neurological examination, higher mental functions and cranial nerves examination was normal but there was hypotonia with diminished reflexes. Other systems examination was normal. Infantogram showed narrow thorax with no dysplastic or dysostosis changes in long bones and spine (Figure 2A and 2B), echocardiography had small PDA and OS ASD with left to right shunt with mildly dilated right atrium and right ventricle. The neonate had persistent respiratory distress and persistent metabolic acidosis and succumbed on day 5. Based on examination findings like flat face, cataract with mild joint laxity there was likely possibility of stickler syndrome but the presence of persistent metabolic acidosis was the point which was against and as the neonate was deteriorating even with ventilatory support, metabolic work up has been done to look for inborn errors of metabolism and blood sample has been stored for genetic evaluation.
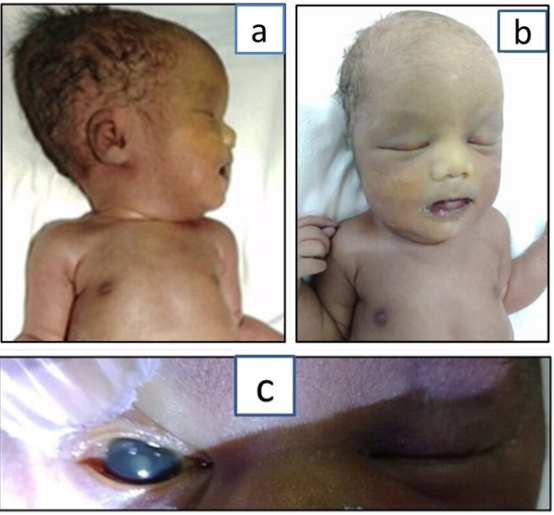
Figure 1: Figure 1A to 1C depicts facial features of the child. 1ANeonate with lateral facial profile. 1B-Frontal facial profile of the neonate. 1C-Congenital cataract.
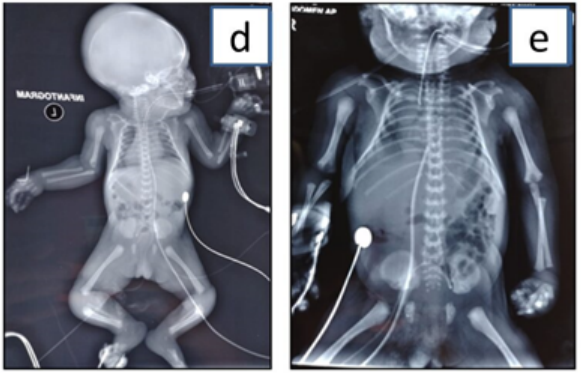
Figure 2: Figure 2A and 2B depicts skeletal features. 2A-Infantogram with normal long bones. 2B-Narrow thorax.
Arterial blood gas analysis (ABG) showed PH of 7.1 (metabolic acidosis), plasma lactate was elevated (9.2 mmols/L). Serum electrolytes, creatine phosphokinase (CPK) were normal. Liver function tests were normal except for reduced proteins. Extended newborn screening was normal. Urine GCMS showed elevated lactate, 3(OH) butyric acid and 2 (OH) butyric acid. Based on the history, examination and basic work up a presumptive diagnosis of inborn error of metabolism most likely a Mitochondrial disorder has been made and clinical exome sequencing of the neonate has been done.
Clinical exome sequencing has shown a likely pathogenic homozygous variant c.580C>T (p.Arg194Cys) [NM_005262] in exon 3 of GFER gene known to cause Mitochondrial progressive myopathy with congenital cataract, hearing loss and developmental delay. This variant was previously reported in a neonate with persistent lactic acidosis and facial dysmorphism. This variant was absent from population databases as well as in-house database. In-silico predictions reported it to be damaging, CADD score was 27.0, and the codon was conserved across mammalian species. This variant was consistent with the presumptive diagnosis of the child.
The possibility of genetic etiology is more likely in neonates with cardio-respiratory insufficiency needing ventilator support for prolonged period, in those with neonatal encephalopathy and intractable seizures not responding to treatment and in neonates with multiple malformations. It is utmost important to have exact etiological diagnosis of these neonates with high risk of mortality to avoid pointless intensive care and for appropriate genetic counseling of the couple for future reproductive choices. Genetic diagnosis of rare genetic disorders on the basis of clinical presentation in critically ill newborns is extremely challenging.
Mitochondrial disorders are the group of clinically heterogeneous multisystem disorders caused due to dysfunction of mitochondrial respiratory chain activity leading to abnormal oxidative phosphorylation and these disorders can manifest anytime from birth to adulthood [1]. Mitochondrial progressive myopathy with congenital cataract, hearing loss and developmental delay (MPMCD) is a rare mitochondrial disorder caused by pathogenic variants in GFER gene and is characterized by abnormalities spanning multiple organ systems. Accumulation of multiple mtDNA deletions leads to progressive myopathy. The Human Phenotype Ontology (HPO) allows composite phenotypes to be represented systematically in such cases.
GFER gene is part of the mammalian iron-sulfur cluster assembly genes which are essential for numerous biological processes, including mitochondrial respiratory chain activity. GFER mutations thus lead to combined respiratory chain deficiency leading to human mitochondrial disorders.
Di Fonzo., et al. first reported MPMCD in a Moroccan family where 3 children were affected with congenital cataract, muscular hypotonia, sensorineural hearing loss, and developmental delay [2].
The first report of MPMCD in a neonate was reported by van Diemen CC., et al. in a 5 day old child with persistent lactic acidosis and facial dysmorphism [3].
Our index case had facial dysmorphism in the form of flat face, corneal clouding, congenital cataract and persistent lactic acidosis, respiratory distress not responding to ventilatory support and hypotonia with diminished reflexes. Narrow chest with brachydactyly, varus deformity of foot and mild joint laxity which were additional findings seen in our case were not reported previously.
Genotype-phenotype correlations are not well established in MPMCD but congenital cataract was common finding in all cases of MPMCD with varying degree of severity in developmental delay [4].
Exome sequencing thus has an important value in the neonatal and pediatric intensive care in addition to routine work up and in addition invasive diagnostic tests such as muscle biopsies can be avoided. Next generation sequencing (NGS) can be further exploited in such cases if detailed phenotyping is integrated with NGS results.
This case highlights that this is the second case report on co-association of Mitochondrial progressive myopathy with congenital cataract, hearing loss and developmental delay diagnosed in a neonate. In addition it also illustrates that next generation sequencing has revolutionized the genetic work-up for the critically ill newborns as accurate diagnosis is an integral component for those with rare genetic disease and in providing appropriate genetic counseling for future reproductive choices.
The authors wish to thank the patient and his family for their cooperation.
None.
The authors declare no conflict of interest.
Copyright: © 2021 Gayatri Nerakh., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.