Maryam Alsada* and Khadija Abdool Abdelbary
Department of Pediatrics, Dubai Hospital, Dubai Health Authority (DHA), United Arab Emirates
*Corresponding Author: Maryam Alsada, Department of Pediatrics Dubai Hospital, Dubai Health Authority (DHA), United Arab Emirates.
Received: February 09, 2021; Published: March 09, 2021
Citation: Maryam Alsada and Khadija Abdool Abdelbary. “X Linked Agammaglobulinemia with a Novel Genetic Mutation in the BTK Gene”. Acta Scientific Paediatrics 4.4 (2021): 20-24.
X-linked agammaglobulinemia (XLA) is an X-linked inherited disease caused by a germline mutation in the BTK gene leading to Bruton’s tyrosine kinase deficiency, which results in failure of B lymphocyte precursors to mature into B lymphocytes and ultimately plasma cells. This in turn will cause severe deficiencies in all types of immunoglobulins and therefore increased susceptibility to various bacteria and viruses. There are many pathogenic variants in the BTK gene that have been linked to XLA, with members in the same family showing heterogeneity.
Here, we describe a case of a 22- month-old boy with XLA who was found to have a Novel genetic mutation in the BTK gene. We will investigate the various mutations described in XLA and genotype-phenotype correlations.
Keywords: Primary Immunodeficiencies (PID); X-Linked Agammaglobulinemia (XLA)
XLA: X linked Agammaglobulinemia; BTK: Bruton’s Tyrosine Kinase; PID: Primary Immunodeficiencies
Primary immunodeficiencies (PID) encompass more than 250 disease entities of different incidence and clinical importance. Primary humoral deficiency syndromes are a group of inherited disorders characterized by an inability to develop clinically effective immunoglobulin production. The most common clinical presentation of PID is recurrent infection and depending on how severe the genotype this may be, the age of presentation and clinical outcomes [1,2]. The first described disease from this group is Xlinked agammaglobulinemia (XLA), which was previously named Bruton’s agammaglobulinemia after Dr. Ogden Bruton. The age of presentation is noted after 3 months of age but less than 1 year of age. More than 90% of affected males present with unusually severe or recurrent sinopulmonary infections. Notably, Recurrent Otitis Media after the age of 12 months, but these patients are not evaluated for immunodeficiency until after their first hospitalization [3,4]. Meningitis, osteomyelitis, sepsis, and GI tract infectious (e.g. gastroenteritis or diarrhea) are less common initial manifestations of XLA [5].
The European society of immunodeficiencies has defined Agammaglobulinemia as Fewer than 2% circulating B cells (CD19 and CD20), preferably in two separate determinations and a normal number of T cells (CD3, CD4 and CD8) AND serum IgG levels below: 200 mg/dl in infants aged < 12 months or 500 mg/dl in children aged > 12 months OR normal IgG levels with IgA and IgM below 2 Standard deviations AND onset of recurrent infections before 5 years of age [6].
The inability to produce immunoglobulins leaves the host susceptible to a wide array of infections, tissue damage, and potentially fatal consequences which under normal circumstances would be undemanding to a functioning immune system.
There are more than 600 different mutations in the BTK gene have been found to cause X-linked agammaglobulinemia (http:// bioinf.uta.fi/BTKbase). Most of these mutations result in the absence of the BTK protein while some can cause a single amino acid, but both of which would have the same result.
Some people with XLA have large DNA deletions that remove one end of the BTK gene and all of a neighboring gene known as TIMM8A. Mutations in TIMM8A cause deafness-dystonia-optic neuronopathy (DDON) syndrome, which is characterized by hearing loss, vision problems, dementia, and dystonia or ataxia. These Individuals present with signs and symptoms of both XLA and DDON syndrome.
A few mutations in the BTK gene have been found to cause isolated growth hormone deficiency type III, a condition characterized by slow growth, short stature, and a weakened immune system. Mutations that cause this condition lead to the production of a nonfunctional version of the BTK protein [6].
The database of mutations for Agammaglobulinemia continues to grow and studies done to look at genotype-phenotype correlations have in themselves come across up with one-third of their study groups having novel mutations [7,8]. The fact that a great number of novel mutations were found in a relatively limited number of patients underlines the heterogeneity of BTK mutations in various populations.
In this way, the knowledge about the causes of human genetic disorders constantly improves with this continuous identification of novel disease genes and mutations.
Our case represents a boy who was diagnosed at 22 months of age with immunodeficiency with a clinical and laboratory diagnosis of pseudomonas sepsis with ecthyma gangrenosum.
Here we had 22 months old boy, known to have G6PD deficiency, who was born at term to non-consanguineous parents with emergency LSCS due to cephalic pelvic disproportion with thick meconium. His birth weight was 4.140Kg. He was vaccinated up to age and had achieved growth and developmental milestones at the appropriate age.
He has no significant family history to note.
A brief Medical history:
At 22 months of age, he was again admitted to the pediatric general ward as a case of bronchopneumonia and sepsis. He presented with a history of upper respiratory tract infection in the form of cough, cold, and high-grade fever of 7 days duration and rash over the lower limbs. He had associated decreased oral intake with 3 days of loose stools and decreased activity. Parents seek medical advice over the course of 7 days during which he received an oral antibiotic course with no improvement.
Upon clinical examination, he was noted to be listless, capillary refill time less than 3 seconds, throat clear, on auscultation of the chest, he was noted to have decreased breath sounds over the left upper lobe with Rhonchi bilaterally, Cardiovascular examination was normal, Neurological examination showed GCS of 14/15 and over his skin there was a notable indurated, erythematous, localized, tender swelling on the left shin significantly increasing in size and another one on the lateral part of the right knee, as a possible cutaneous manifestation of Sepsis. He was started on Empirical Antibiotics and urgent workup was done including Lumbar puncture.
Blood investigations were collected and Lumbar puncture was done which yielded high inflammatory markers and his blood culture grown Pseudomonas aeruginosa. Lumbar puncture showed CSF WBC 277/cmm with normal protein and so he was managed as a case of pseudomonas meningitis with sepsis and Bronchopneumonia. Over the course of 2 days, the indurated lesions over the lower limbs rapidly progressed to Ecthyma Gangrenosum (Figure 1).
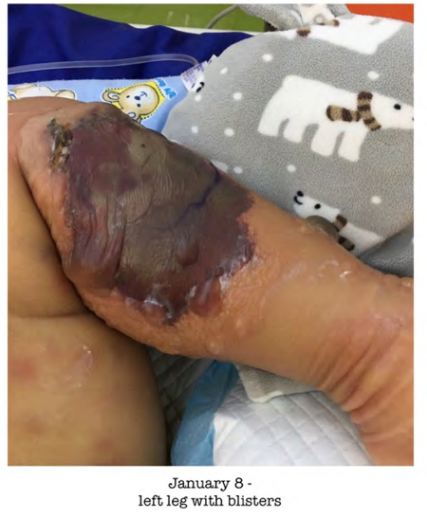
Figure 1: Left lower limb with skin lesions which clinically correlate with of ecthyma gangrenosum.
His antibiotics were changed as per culture sensitivity to IV ceftazidime and gentamycin. He received IV ceftazidime and gentamycin for 15 days but he remained febrile. Again, a full septic workup has been collected for him. Chest X ray which showed diffuse haziness, blood culture, central and peripheral line culture, fungal blood culture, and lumbar puncture were repeated including enterovirus and herpes virus which all came negative. Antibiotic was changed from gentamicin (after a course of 15 days) to Tazocin while Ceftazidime was continued. Ceftazidime was stopped after completing a course of 32 days and Tazocin was continued for total of 4 weeks after which he made a full recovery.
Regarding his skin lesions, there were two forms of lesions. One in the form of palpable maculopapular and nodular lesions blanching, that has spread all over the body. The second type were bullae mainly over the lower limbs, which increasing in size and number. The largest was over the popliteal aspect of the lower limbs were increasing in size and number in the lower limbs. Our Dermatologists were consulted and advised to aspirate the Bullae. These lesions correlated with Ecthyma gangrenosum, which is usually seen in pseudomonas sepsis in immunocompromised individuals. Pediatric surgery and Plastic surgery teams were consulted regarding the wound care of the skin lesions. Bed side debridement by the plastic surgeons was done twice and followed by dressing and care by the wound care team. During the first debridement swabs were collected for wound culture from both limbs, which was negative on the left side and positive for pseudomonas on the right side.
After 7 days, second debridement was done, wound culture was repeated for both sites, which came negative. After 6 days of second debridement, skin Grafting was done. Skin graft was then followed up by the plastic surgery team and wound care team. Before discharge, the plastic surgeon did the final dressing and reviewed the graft and gave the family education about how to take care of the wound at home and the follow-up plan.
This presentation along with his past medical history raised the suspicion of possible immunodeficiency for which immune status, immunoglobulins levels, measles, and mumps antibodies were sent. Immune status assay showed Total absence of NK and B cells are suggestive of Severe combined immunodeficiency (SCID). Measles and mumps antibodies were negative although he had received MMR vaccination at the age of 12 months.
Whole exome sequencing was later sent which showed that he had a pathogenic variant mutation of the BTK gene which has been linked to X linked Agammaglobulinemia. This particular splice site variant of the BTK gene has never been documented or annotated in any genetic database so far (See table 1).

Table 1: Description of a novel mutation in BTK gene.
Accordingly, he had received 2 doses of IVIG along with completed the course of antibiotics after which he showed gradual overall improvement in his overall clinical condition.
He was discharged from our ward in good condition after almost 2 months of hospitalization. His Parents were oriented and educated about the nature of the disease and how to take daily life precautions for him and he is planned to be on regular IV human immunoglobulins, 0.5 gm/kg every 4 weeks.
X-linked agammaglobulinemia is one of the most clinically important deficiencies of humoral immunity. The disease is caused by mutation in the BTK gene (Xq21.3-Xq22), which leads to deficiencies in terminally differentiated plasma cells. This molecule is essential in the signaling through B-cell receptor [9].
Affected individuals have markedly reduced levels of all major classes of immunoglobulins in serum and present an increased susceptibility to severe and recurrent bacterial infections from early childhood and according to the severity of the gene mutation may be the age of onset and severity of infections.
In our case, a 22-month-old boy presented clinical sepsis, culture negative at 4 days of life which he responded well to antibiotics and again at 11 months of age with febrile neutropenia suspected to be secondary to viral infection after which he made a full recovery within a week. In between these episodes up until 22 months of age, he continued to thrive well and was stable. At 22 months of age, he presented with pseudomonas sepsis and associated Ecthyma gangrenosum on the lower limbs. Our patient had no significant family history of suspected or confirmed immunodeficiencies, only had 2 previous admissions, and was well in between admissions. In view of the current presentation, immune work-up was done and he was then diagnosed with X linked Agammaglobulinemia with a novel mutation.
Approximately two-thirds of the patients have a familiar mutation, in one-third a new mutation has been identified with no single mutation that accounts for more than 5% of patients. It is very important, that despite some general common symptoms of XLA, the genotype-phenotype correlation in XLA is still said to be weak [2,10]. With rapidly evolving diagnostic modalities, reanalysis of new mutations and screening of unaffected family members may yield more insight into whether there is a genotype-phenotype association as well as identification of further genetic causes. The importance as physicians is to determine the underlying mutations to anticipate possible clinical presentation and catch them in early stages to effectively initiate early treatment, thereby minimizing the effects on neurodevelopmental outcomes, morbidity, and better quality of life at later stages as well as proper anticipatory genetic counselling.
This project required no specific support with regards to funding. The authors have no financial relationships relevant to this article to disclose.
The authors have no conflicts of interest to disclose.
Copyright: © 2021 Maryam Alsada and Khadija Abdool Abdelbary. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.