Srijan Singh*
Senior Registrar, Department of Neonatology, Seth G S Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India
*Corresponding Author: Srijan Singh, Senior Registrar, Department of Neonatology, Seth G S Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India.
Received: December 11, 2020; Published: January 22, 2021
Citation: Srijan Singh. “A Case of Early-infantile Epileptic Encephalopathy with Suppression-bursts- the Ohtahara Syndrome”. Acta Scientific Paediatrics 4.2 (2021): 06-10.
Ohtahara syndrome (OS) is an epileptic syndrome with onset in neonatal period and has many clinicoelectrical characteristics, of which age dependency and evolutional change is specific. The most specific EEG feature is the suppression burst (SB). This pattern is characterized by high voltage bursts alternating with flat suppression phases at a regular rate. A term neonate presented with seizures on day three of life. Seizures were intractable. Metabolic workup for seizures was normal. Extended panel neurometabolic screen was normal and neuroimaging did not reveal any structural abnormality. EEG showed burst suppression pattern suggestive of ohtahara syndrome. Virtually all reported cases of early infantile epileptic encephalopathy (EIEE) are secondary to a congenital or acquired structural malformation of cortical development. The etiology of this syndrome remains obscure. All children with EIEE should be thoroughly investigated with MRI, CSF amino acid level determination, and detailed postmortem neuropathologic examination.
Keywords: Ohtahara Syndrome; Seizures; Epileptic Encephalopathy; EEG; Suppression Burst
OS: Ohtahara Syndrome; EEG: Electroencephalography; STXBP1: Syntaxin-binding Protein 1; SNARE: Soluble N-Ethylmaleimide Sensitive Factor Attachment Protein Receptors; LGS: Lennox Gastaut Syndrome; WS: West Syndrome; EIEE: Early Infantile Epileptic Encephalopathy; MRI: Magnetic Resonance Imaging; CSF: Cerebrospinal Fluid; GABA: Gamma-aminobutyric Acid; NICU: Neonatal Intensive Care Unit.
Ohtahara syndrome (OS) is an epileptic syndrome with onset in neonatal period and has many clinicoelectrical characteristics, the youngest form in the frame of age dependent epileptic encephalopathies. The age-dependent epileptic encephalopathy, proposed by Ohtahara, has the following characteristics:
There are four special features for adopting the term “epileptic encephalopathy” instead of “epilepsy”: (1) presence of underlying disorders, (2) extremely frequent seizures, (3) continuously and diffusely appearing epileptic abnormality on electroencephalography (EEG) (4) mental deterioration manifesting with the persistence of seizures. These tonic spasms resemble West Syndrome (WS), but have some differences; appearance in both waking and sleeping states and no clustering. Daily seizure frequency is very high, ranging from about 100 to 300 times in those with isolated seizures, and from 10 to 20 series in those with seizures in series. Most specific EEG feature is the suppression burst (SB) which is a pattern characterized by high voltage bursts alternating with flat suppression phases at a regular rate. Regularity of periodicity and bursts containing randomly appearing frequent multifocal epileptic discharges distinguish this pattern from burst-suppression, which is a non-specific EEG abnormality observed in newborns with severe neurological insult. Structural abnormalities of the brain are the main cause of OS. It is a static encephalopathy rather than a metabolic encephalopathy.
Syntaxin-binding protein 1 (STXBP1) (also known as MUNC181) is a protein of the SEC1 family of membrane trafficking proteins predominantly expressed in brain, which plays an important role in synaptic vesicle docking and fusion. Through interaction with both vesicle-associated (synaptobrevin 2 or vesicle-associated membrane protein 2) and target associated (syntaxin-1 and synaptosomal-associated protein 25) soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNARE) proteins, STXBP1 modulates the presynaptic vesicular fusion reaction. STXBP1 is encoded by the STXBP1 gene, consisting of 20 exons and located on chromosome 9q34.11 [2].
Seizures are intractable and prognosis is grave with severe psychomotor retardation and deaths in early infancy. Hence, it is known as catastrophic epilepsy with other forms of the age-dependent epileptic encephalopathy, i.e. West syndrome and Lennox Gastaut Syndrome (LGS). Clonazepam and acetazolamide are effective in some cases. In addition, high dose vitamin B6, valproate, vigabatrin or ketogenic diet should be tried, but will be mostly ineffective.
The patient was born to a 37 year old G3P1L1A1 mother by caesarean section at term gestation. Antenatal scans were normal. Baby’s mother did not have any medical or surgical illnesses. There was no family history of seizures. Baby cried immediately after birth. Birth weight was 1985 grams and was started on breastfeeds. On day three of life, the neonate was noted to have tonic seizures with shrill cry (Figure 1).
Blood glucose levels were normal. Sepsis screen and lumbar puncture were normal. MRI Brain did not reveal any structural abnormality (Figure 2). Calcium levels were low. Calcium supplementation was done after which levels normalized. Initially, phenobarbitone loading was done. Levetiracetam, pyridoxine and vitamin B12 were added later. But seizures continued. Seizures were frequent and not accompanied by autonomic phenomena, grunting respiration or colour change. Seizures occurred when the babies were awake or asleep, but showed state dependency, more frequent when the babies were disturbed or awakened. Following onset of seizures, infants became inactive and hypotonic, lost visual alertness and required nasogastric or intravenous feeding. Tendon reflexes were preserved. Baby was then shifted to our NICU for further management. On evaluation, glucose, calcium and magnesium levels were normal. Sepsis screen was negative and repeat lumbar puncture was normal. MRI did not reveal any structural abnormality. Newborn screening reports were normal. Extended panel neurometabolic screening was normal. Serum ammonia levels were borderline raised. EEG showed suppression burst pattern suggestive of ohtahara syndrome (Figure 3). Baby was noted to have multiple seizures- tonic seizures, subtle seizures and multifocal clonic seizures. The neonate received maintenance phenytoin, levetiracetam, valproate and a trial of pyridoxine, biotin and pyridoxal phosphate but continued to have intermittent seizures. Seizures were finally controlled on topiramate. Neurological examination was abnormal at the time of discharge and the neonate had axial and appendicular hypotonia. Early intervention therapy was started. Baby did not have seizures on follow up at the age of three months and is on levetiracetam and topiramate. This is a rare case of OS diagnosed in the neonatal period presenting with intractable seizures. This case is unique because seizures were responsive to topiramate which blocks voltage-dependent sodium and calcium channels. It also inhibits the excitatory glutamate pathway while enhancing the inhibitory effect of GABA.
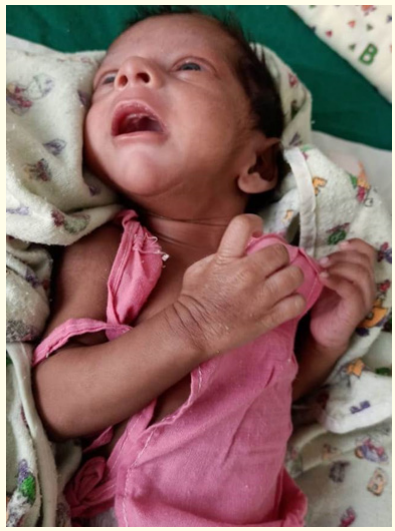
Figure 1: Neonate with ohtahara syndrome.
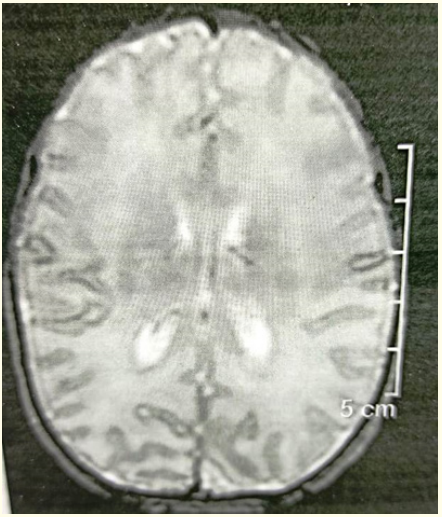
Figure 2: MRI brain of the patient.
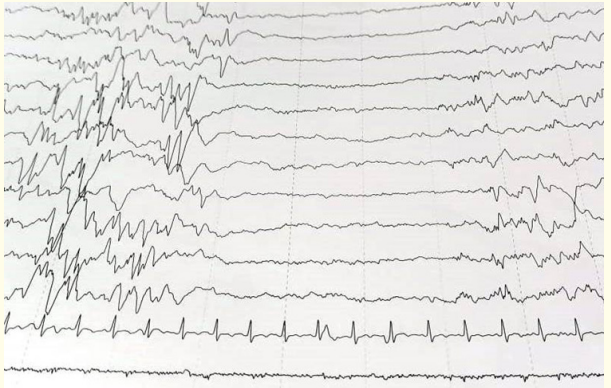
Figure 3: EEG of the neonate showing suppression burst pattern.
Clarke et al described identified eleven infants with the clinical and electroencephalographic features of early infantile epileptic encephalopathy (EIEE). Onset of seizures was on the first day of life for seven babies and fifth day for the remainder. Tonic seizures (either generalized or lateralized and often in series) were the most frequent type. Seizures were frequent and accompanied by excess salivation, grunting and colour change. Seizures showed state dependency, more frequent when the babies were disturbed or awakened. Following onset of seizures the infants became inactive and hypotonic, lost visual alertness and required nasogastric or intravenous feeding. Tendon reflexes were preserved or increased. Seizures were intractable throughout the neonatal period. Ten infants had interictal recordings in the neonatal period, and all showed suppression burst, high-voltage paroxysmal activity separated by periods of no recognizable activity which lasted for up to 18 seconds. EEGs of two infants showed prolonged periods of unilateral suppression burst. These recordings showed recruitment of lateralized rhythmical activity becoming more generalized and increasing in frequency and amplitude as the seizure progressed, and decreasing in frequency and amplitude as the clinical attack subsided. The eleven babies described had neonatal onset of intractable seizures, showed characteristic EEG findings and a very poor outcome. No cause could be found for eight [3]. In our case, patient developed seizures on day three of life. Seizures were more frequent in the awake state. EEG showed burst suppression pattern. MRI brain did not reveal any abnormality.
Miller and colleagues described a case of early infantile epileptic encephalopathy (EIEE) with suppression-bursts (Ohtahara syndrome) associated with a diffuse cerebral migrational and maturation disorder. Onset of seizures was on day one of life and characterized by generalized tonic and clonic movements. Electroencephalogram (EEG) at 2 days of age revealed a continuous disturbance of cerebral activity with a suppression-burst pattern and electrographic seizures. Magnetic resonance imaging (MRI) at 1 month of age revealed delayed myelination and a thin corpus callosum. Neurometabolic evaluation was normal. CSF lactate was normal. CSF amino acid profile showed undetectable gamma-aminobutyric acid (GABA). Postmortem pathologic examination was suggestive of a cerebral migration and maturation disorder with diffuse subtle microdysgenesis of the cerebral cortex. There was bilateral enlargement of the dentate nuclei with loss of the normal ribbon undulation and a thick multinodular appearance [4]. This is similar to our case where MRI brain did not reveal any significant abnormality. There might exist an underlying cortical laminar disorganization corresponding to an insult in the late embryologic stage of migration and organization as in this case study.
Yamatogi and colleagues studied sixteen cases (9 males and 7 females) of OS. No obvious sex difference was observed. Ages at their first visit to us ranged from 11 days to 4 months 7 days. Twelve of the cases (75%) had their onset within 1 month and all had tonic spasms. Neuroimaging revealed structural abnormalities suggestive of two cases of Aicardi syndrome, one case each of hemimegalencephaly, porencephaly, hydrocephalus, lissencephaly, and four cases of non-specific cerebral atrophy or dysgenesis. These were classifiable into two groups: group I of seven cases evolving from suppression burst to hypsarrhythmia and further to diffuse slow spike-waves, and group II of nine cases demonstrating spike foci. All seven cases died in group I, but only one of the nine cases died and seven cases were seizure free in group II [5]. In our case, baby was noted to have multiple seizures- tonic seizures, subtle seizures and multifocal clonic seizures. MRI Brain was normal. Baby has to be followed up with EEG to see for evolution to WS or LGS.
Almost all of the reported cases of EIEE are secondary to a congenital or acquired structural malformation of cortical development. Etiology of this syndrome still remains obscure. The role of GABA in EIEE and in epileptogenicity of malformations of cortical development requires further investigation to elucidate causal mechanisms.
All children with EIEE should be thoroughly investigated with MRI, CSF amino acid level determination, and detailed postmortem neuropathologic examination.
The author expresses sincere gratitude to all the neonatology unit staff and all the colleagues in the pediatric neurology and radiology unit at the Seth G S Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India.
The authors declare that they have no competing interest.
Copyright: © 2021 Srijan Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.