Nagamani Agarwal*
Professor, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India
*Corresponding Author: Nagamani Agarwal, Professor, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India.
Received: July 27, 2020; Published: September 16, 2020
Citation: Nagamani Agarwal. “Antenatal Barter Syndrome - Novel Mutation in Gene SLC21A1”. Acta Scientific Paediatrics 3.10 (2020): 17-20.
Bartter’s Syndrome (BS) is a rare inherited renal tubular disorder characterized by hypokalemia, hypochloremic metabolic alkalosis and urinary wasting of sodium, potassium and chloride. It is caused by mutations in several genes encoding for ion transporters in the thick ascending limb of loop of Henle.
Antenatal BS (type I) is caused by mutation in the SLC12A1 gene, encoding for sodium-potassium-2-chloride- cotransporter (NKCC2). This patient was born prematurely with low birth weight and mother underwent therapeutic amniocentesis after 24 weeks of pregnancy for severe polyhydramnios. Child was evaluated at 7 months of age for failure to thrive, developmental delay in the motor domain and polyuria from early infancy. Investigations favored the diagnosis of BS.
Genetic study revealed presence of homozygous mutation (p.Cys475Arg variant) in exon 11 of the SLC12A1 gene on chromosome 15. This homozygous mutation, not reported in the literature so far may be a novel one in this family.
Keywords: Antenatal Bartter’s Syndrome; Hypokalemia; Mutation
Bartter’s syndrome (BS), first described in 1962 by Frederic Bartter, is a rare hereditary hypokalemic salt losing tubulopathy (SLT) [1]. It is characterized by hypokalemia, hypochloremic metabolic alkalosis, normal blood pressure and urinary wasting of potassium, sodium and chloride with varying degree of hypercalciuria. Clinical disease results from defective tubular reabsorption of sodium and chloride in the thick ascending limb (TAL) of loop of Henle, where normally 30% of filtered salt is reabsorbed. Key player of active sodium and chloride uptake into tubular cells in TAL is the apically expressed furosemide sensitive sodium- potassium -2-chloride- cotransporter (NKCC2). Gene SLC12A1 codes for NKCC2. Mutation in this gene results in defective reabsorption of salt in TAL.
This is a case report of antenatal BS due to homozygous mutation (p.Cys475Arg variant) in exon 11 of the gene SLC12A1. This homozygous mutation, not reported in the literature so far may be a novel one in this family.
An eight month old female infant born late preterm with low birth weight (2.2 kg) as 3rd issue of consanguineous marriage presented with complaints of failure to gain weight, developmental delay predominantly in the motor domain, polyuria, polydipsia and constipation since 3 months of age. Polyhydramnios was detected antenatally and therapeutic amniocentesis was done 3 times after 24 weeks of pregnancy.
Mother had polyhydramnios in previous two pregnancies. First pregnancy resulted in intrauterine death at 7 months. Female child, born from 2nd pregnancy had similar complaints of polyuria, failure to thrive and died at 14 months of age.
Examination revealed a wasted and stunted child with weight and length less than 3rd centile on WHO growth charts. Blood pressure was within normal limits. There was hypotonia in all the extremities. Rest of systemic examination was normal. Investigations are shown in the table 1.
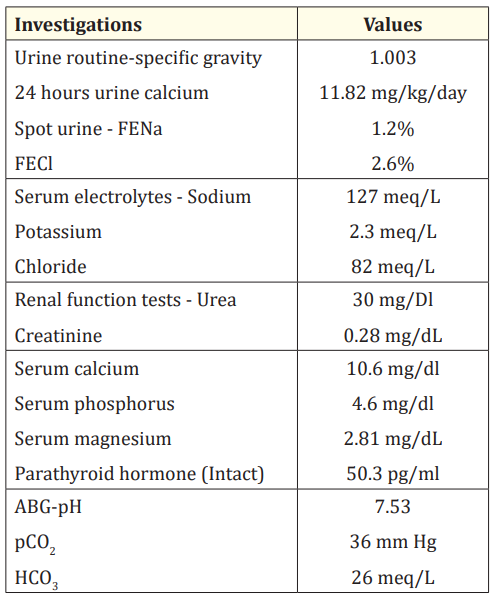
Table 1: Urine and serum biochemical profile.
USG abdomen showed normal sized kidneys with increased echogenicity of renal parenchyma. Hyperechoic foci were noted in both the renal medulla suggestive of nephroclacinosis. Bilateral mild pelviectasia was noted.
Based on the aforesaid clinical features and investigations (hypokalemia, hypochloremic metabolic alkalosis, increased urinary chloride, hypercalciuria and nephrocalcinosis) diagnosis of antenatal Bartter’s syndrome was considered.
Genetic study (clinical exome sequencing) revealed the presence of homozygous mutation (p.Cys475Arg variant) in exon 11 of the SLC12A1 gene which codes for NKCC2. This Cys475Arg variation results in the amino acid substitution of arginine for cysteine at codon 475. This identified variant is not reported till date in the literature.
Mutation in the SLC12A1 gene results in impaired salt reabsorption in TAL. Since the clinical presentation of this infant is similar to BS, this mutation (p.Cys475Arg variant) is most likely to be the disease causing variant in the family.
Child was started on ibuprofen and potassium supplement following which child showed significant decrease in polyuria and remarkable improvement in motor abilities.
Bartter’s Syndrome is a rare hereditary hypokalemic SLT with a prevalence of 1 in 100,000 population [2]. It is characterized by hypokalemia, hypochloremic metabolic alkalosis and hyperaldosteronism with normal blood pressure and urinary wasting of sodium, potassium and chloride.
In TAL of loop of Henle, transepithelial sodium chloride absorption is facilitated through coordinated activity of apical furosemide sensitive NKCC2, renal outer membrane potassium (ROMK) channel, basolateral NA+/K+ATPase and basolateral CIC-Ka (chloride channel -kidney a) and CIC-Kb (chloride channel - kidney b) ion transporters. NKCC2 is a key player in sodium chloride absorption in TAL and potassium is recycled through apical ROMK channels into the tubular lumen. Calcium and magnesium are reabsorbed passively via paracellular pathway driven by lumen positive transepithelial potential.
Patients with BS have defective transepithelial transport of sodium and chloride in TAL of Henle’s loop due to mutation in genes coding for any of the above ion transporters. This leads to increased delivery of sodium and chloride to distal tubules resulting in salt wasting, hypokalemia and polyuria. The resultant volume depletion causes activation of renin-angiotensin-aldosterone system (RAAS) and subsequent secondary hyperaldosteronism [3]. Long term stimulation causes hyperplasia of juxtaglomerular apparatus and hence increased renin levels. Contraction of ECF is also accompanied by markedly elevated renal and extra renal PGE2 production (hence the term Hyper prostaglandin E syndrome).
Aldosterone enhances sodium absorption in distal nephron by up regulation of E Nac (epithelium sodium channel) and Na-KATPase. This increases sodium reabsorption and potassium and hydrogen secretion resulting in hypokalemic alkalosis. Loss of chloride along with ammonium and potassium causes hypochloremia. Hypercalciuria is typical for loop disorders as paracellular reabsorption of calcium is impaired.
According to pathophysiology, SLTs are classified into two major groups basing on nephron segment directly involved: furosemide like loop disorders (L type) and thiazide like DCT disorders (D-type). Genetically loop disorders are referred to as BS and DCT disorders as Gitelman’s syndrome. Combined loop and DCT dysfunction forms a third group called furosemide -thiazide like SLT (L-DC type). Table 2 depicts the detail classification of SLT basing on location of affected gene and primarily affected nephron segments [4].
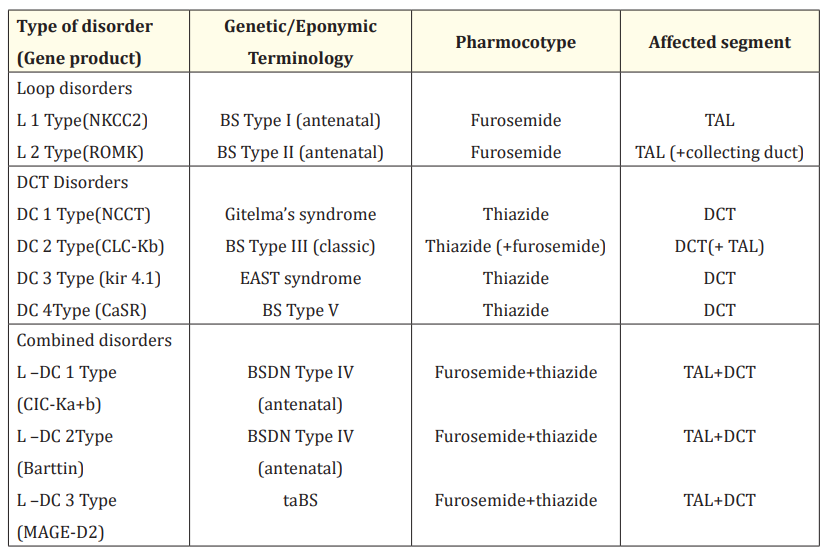
Table 2: Physiologic and pharmacologic classification of Salt losing tubulopathy.
EAST Syndrome: Epilepsy, Ataxia, Sensorineural Deafness and Tubulopathy.
BSDN: Bartter’s Syndrome with Sensorineural Deafness.
taBS: Transient Antenatal Bartter Syndrome.
MAGE-2: Melanoma associated antigen D2.
All types of SLT share common findings of hypokalemia and hypochloremic metabolic alkalosis. Loop disorders (defects in NKCC2, ROMK) and combined defects (defect in Barttin or both CIC-Ka and CIC-Kb) present antenatally as fetal polyuria resulting in maternal polyhydramnios and prematurity. During neonatal period babies may have profound polyuria, recurrent vomiting, life threatening dehydration and electrolyte imbalance. Failure to thrive, recurrent dehydration, polyuria and polydipsia are common presentations in infancy.
Nearly all the patients with NKCC2 or ROMK defects develop medullary nephrocalcinosis within first few months of life due to persistently high urinary calcium excretion.
Our patient presented with classical clinical and biochemical features of Antenatal BS. Child showed medullary nephrocalcinosis and hypercalciuria. Association of antenatal BS with hyperparathyroidism has been observed in some cases [5,6]. However, paratharmone (intact) level in plasma was in the normal range.
Genotyping to detect mutation in the disease causing gene was done in view of suspicion of two elder sibs being affected with similar problem. Many mutations in each of the 5 genes causing different types of BS have been identified with effects on proteins ranging from slight compositional and functional changes to complete deletion [7].
Clinical exome sequencing revealed the presence of patient homozygous mutation (p.Cys475Arg variant) in exon 11 of the gene SLC12A1 on chromosome 15. Gene SLCI2A1 codes for ion cotrasporter NKCC2 and mutations in this gene causes BS - type I. Since the clinical presentation and laboratory investigations favored the diagnosis of BS in our patient, this mutation can be the most likely disease causing variant and diagnosis of type I BS can be made. This identified variant is not reported till date in any literature or database and may be a novel one in this family. Genetic study has been advised for both the parents to detect if they are carriers of the same mutation.
Following treatment with ibuprofen and potassium supplements, child showed remarkable improvement in her motor abilities and significant reduction in polyuria.
Copyright: © 2020 Nagamani Agarwal. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.