Kavitha Pallad1, Venkatraman Bhat2* and Suhel Hassan1
1Department of Otolaryngology, Mazumdar Shaw Medical Center, Narayana Health, Bangalore, India
2Department of Radiology, Mazumdar Shaw Medical Center, Narayana Health, Bangalore, India
*Corresponding Author: Venkatraman Bhat, Department of Radiology, Mazumdar Shaw Medical Center, Narayana Health, Bangalore, India.
Received: October 03, 2019; Published: October 23, 2019
Citation: Venkatraman Bhat., et al. “Congenital Aural Atresia with a Cholesteatoma – Report on the Management of a Rare Case and Review of Literature”. Acta Scientific Paediatrics 2.11 (2019):55-58.
Congenital cholesteatoma in association with aural atresia in an adolescent is a rare entity in clinical practice. We present a case of congenital aural atresia associated with a cholesteatoma (type D, Schuknecht) managed surgically with successful outcome. The challenge for the surgeon was to create a functional conductive pathway from the external ear to the cochlear apparatus, preserving the facial nerve and labyrinth function. Tympano-mastoidectomy was performed with successful outcome. Improved hearing was evident clinically, subsequently documented with pre-operative and post-operative pure tone audiometry. Brief review of embryology, classification of the entity and management are presented.
Keywords: Cholesteatoma; Congenital Aural Atresia; Tympano-mastoidectomy; HRCT Temporal Bone; Computed Tomography
EAC: External Auditory Canal; HRCT: High Resolution Computed Tomography.
Congenital aural atresia is characterized by hypoplasia of the external auditory canal, often in association with dysmorphic features of the auricle, middle ear and occasionally the inner ear structures [1]. It is relatively rare, occurring in 1.5 in 10,000 to 15,000 births. It has male predominance and occurs bilaterally in 25% [2]. It may be a part of syndromes such as Treacher-Collins syndrome, Crouzon’s syndrome, Klippel-Feil syndrome, Pierre Robin syndrome and Goldenhar syndrome [3,4]. Severity of the condition varies according to the extent of malformation. Detailed anatomical information can be demonstrated and stratified in imaging studies. Congenital cholesteatoma has been reported in about 4-7% of cases of congenital external auditory canal (EAC) atresia [4].
Abnormalities of the facial nerve like dehiscence of the fallopian canal and abnormal course are common in aural atresia. The inner ear development is mostly normal. The usual presenting symptom is ear deformity followed by unilateral or bilateral conductive hearing loss, ranging from 45 to 60 dB in complete atresia and from 30 to 40 dB in partial atresia. High resolution computed tomography (HRCT) of the temporal bone is essential to evaluate the status of the middle ear ossicles, facial nerve anatomy, mastoid pneumatisation and associated anomalies. The main aim of the otologist is the restoration of the hearing of the patient through appropriate surgical options.
A 17-year-old female presented with history of discharge from the left ear on and off and decreased hearing in the same ear since childhood. On examination she had a shallow external auditory canal with a large external auditory canal polyp. The tympanic membrane was not visualized. The pinna was normal. Tuning fork tests showed conductive hearing loss on the left side and vestibular function tests were normal. Pure tone audiometry showed hearing loss about 73 db on the left side. Examination of the right ear was normal.
HRCT of the temporal bone demonstrated a shallow left EAC ending with an atretic plate of 8mm thickness. Tympanic cavity was small with no ossicles (Figure 1). A soft tissue density was seen occupying the remaining middle ear space and mastoid cavity. Inner ear structures were normal. Facial nerve showed an aberrant, somewhat anterior course. Right ear structures were normal. Based on these observations, diagnosis of aural atresia with ossicular, facial nerve anomaly (Type D) in association with cholesteatoma was made. In order to remove the cholesteatoma and restore hearing pathway, tympano-mastoidectomy was considered.
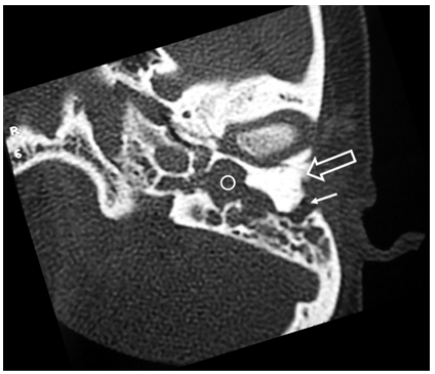
Figure 1: Axial CT image at the level of the left external and middle ear demonstrate large thick atresia plate (open arrow). External auditory canal is a small (white arrow). Middle ear cavity shows soft tissue density (circle). There are no ossicles in the middle ear. There is poor mastoid pneumatisation.
Under general anaesthesia tympano-mastoidectomy was performed and cholesteatoma was removed (Figure 2a, 2b). On exploration no ossicles were seen. The facial nerve canal was dehiscent and visualised far more anteriorly and inferiorly. Dehiscent facial canal was covered with temporalis fascia graft followed by meatoplasty. Post-operative period, which lasted 4 days, was uneventful. Follow up examination after two months showed well epithelised cavity. There was an improvement in hearing with audiometry response at 50 db.

Figure 2: Surgical photographs (a) demonstrate soft tissues representing cholesteatoma in the region of the external auditory canal (open arrow). (b) View of the middle ear cavity after clearing the cholesteatoma and fluid in the middle ear. No ossicles were present.
The inner ear, middle ear and external ear develop independently and often deformity of one does not necessarily be associated with other. Fact is important in conditions like aural atresia where demonstration of normal inner ear provides options for surgical correction of rest of the structures. Abnormalities of the outer and middle ear are often encountered in combination with a normal inner ear structure as the inner ear structures develop from the auditory placode at an earlier during development. Such anomalies occur with frequencies of 11%-30% [5,6]. Invagination of the ectodermal element of first pharyngeal pouch forms EAC. It grows medially to meet the endoderm of middle-ear. The mesodermal component gets entrapped between these ventral and dorsal sites giving rise to meatal plug or plate. Meatal plate undergoes resorption by the end of 28th week of gestation and the external auditory canal undergoes recanalization. Failure of recanalization leads to congenital aural atresia with the formation of atretic plate [5,7].
Various classifications have been proposed for malformations of pinna and EAC used for assessing the severity of the disease. All classifications take in to account of development of EAC, middle ear and mastoids. EAC malformations are classified in to three types [8] or in to four types [1]. The closely interrelated development of the EAC and the middle ear lead to the classification of the combined malformation termed as atresia auris congenita by Altmann [9]. He also described the degree of severity. Additional grading system, based on the set of anatomical information visualised on high resolution computed tomography is proposed by Yeakley and Jahrsdoerfer defining specific quantifiable parameters for prognosis [10].
EAC atresia could be fibrous, osseous or mixed type and middle ear anomalies may accompany the atresia. Congenital aural stenosis predisposes to canal cholesteatoma formation. Extent of cholesteatoma is directly proportional to the stenosis of canal. Cholesteatoma is not seen when EAC width is greater than 4mm. Surgery is advisable in cases showing stenosis less than 2 mm [4].
Congenital cholesteatoma, not associated with aural atresia may be seen at cerebello-pontine angle, petrous pyramid, jugular fossa, middle-ear cavity and mastoid antrum. Proposed mechanisms of pathogenesis are inclusion, migration, or invasion of squamous epithelium, epithelial rests from faulty embryogenesis, or metaplasia of normal epithelium [11]. It is usually asymptomatic, may presents with complications such as otitis media, facial paralysis, hearing loss. Usual pathogens in the infection are pseudomonas aeruginosa, and bacillus proteus spared via Eustachian tube [12]. Our patient with a type D (Schuknecht), is the most severe type and are not considered suitable candidates for repair [1]. There are no reported literature on the exact incidence of this type of anomaly. There are no clear guidelines on surgical management. We opted for surgical option in our patient mainly for relief of recurrent infection secondary to cholesteatoma.
Patients with aural atresia should be evaluated for any syndromic association. They should also undergo oto-acoustic emission and brainstem evoked response audiometry as part of inner ear screening. An HRCT with reconstructions in coronal and axial planes is mandatory for proper evaluation and visualisation of anomalies needed for surgical planning [6] and grading the outcome of the surgery. Contrast enhanced CT study or magnetic resonance imaging (MRI) studies provide detailed information regarding the vascular anomalies and other associated intracranial anomalies, infective complications and presence of congenital cholesteatoma.
HRCT and MRI also provides vital information regarding the facial nerve anatomy and status of the vestibulo-cochlear apparatus. The four important elements in the imaging evaluation are the degree of pneumatisation of the temporal bone, the course of the horizontal and vertical segments of the facial nerve, the existence of the oval window- stapes footplate and the status of the inner ear. CT optimally provides information on the thickness, structure of the bony atretic plate, the size and status of the middle-ear cavity, the soft-tissue component of atresia, and the presence of congenital cholesteatoma.
The two essential requirements for elective surgery in congenital aural atresia are imaging evidence of normal inner ear and audiometric evidence of cochlear function. Surgery in unilateral external auditory canal atresia should be avoided till adolescence or adulthood when patient is able to take the decision on the morbidities of a radical surgery. Generally patients with good pneumatisation of mastoid, normal oval window had good outcome [1,10]. The focus is on clearance of the disease process and then establishing a functional middle ear cavity. Rarely a second look surgery may be required. Surgical transmastoid approach is presently almost obsolete.
Congenital aural atresia (type D) with congenital cholesteatoma, presenting in an adolescent age-group is a rare presentation. It may be associated with anomalous course of facial nerve and canal dehiscence. HRCT of temporal bone is essential for confirming the diagnosis and staging the disease. Documentation of course of the facial nerve is of great importance in planning and performing surgery, thus improving surgical outcome.
Authors would like to acknowledge the efforts of Dr Annapandian V M, in the preparation and publication of this article.
Nil.
Copyright: © 2019 Venkatraman Bhat., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.