Narayanan MP*
Assistant Professor, Department of Biochemistry, Educare Institute of Dental Sciences, KUHS, Kerala, India
*Corresponding Author: Narayanan MP, Assistant Professor, Department of Biochemistry, Educare Institute of Dental Sciences, KUHS, Kerala, India.
Received: June 26, 2019 Published: July 18, 2019
Citation: Narayanan MP. “Isovaleric Aciduria a Case Report of Two Patients”. Acta Scientific Paediatrics 2.8 (2019):45-48.
Isovaleric aciduria (MIM 243500) is known as one of the major organic acidurias and is caused by a genetic deficiency of isovaleryl-CoA dehydrogenase (IVD: E.C.1.3.99.10) catalyzing the third step in leucine catabolism. IVA is clinically characterized by lethargy, vomiting, and an odor of “sweaty feet”; biochemically, IVA is characterized by accumulation of isovaleryl-CoA derivatives that are associated with hyperammonemia and ketoacidosis. IVA can be diagnosed pre-symptomatically as a result of the implementation of newborn screening by tandem mass spectrometry. We report two patients from the same ethnic group who presented with vomiting, seizures, hyperglycemia, metabolic acidosis, increased anion gap and ketosis. One was acute neonatal presentation and the other was chronic intermittent form. Both acute neonatal presentation and the chronic intermittent form can occur in the same ethnic group; suggesting the molecular heterogeneity in the population.
Keywords: Isovaleric Acidemia; Isovaleryl-CoA Dehydrogenase; Newborn Screening; Ketoacidosis; High Performance Liquid Chromatography; Tandem Mass Spectrometry
IVA: Isovaleric Acidemia; IVD: Isovaleryl-CoA Dehydrogenase; NBS: Newborn Screening; HPLC: High-Performance Liquid-Chromatography; GCMS: Gas Chromatography Mass Spectrometry
Isovaleric aciduria has an incidence of 1:250,000 in USA. Deficiency of IVD results in an accumulation of derivatives of isovaleryl-coenzyme A, such as isovaleric acid, 3-hydroxyisovaleric acid, 4-hydroxyisovaleric isovaleryl carnitine and isovaleryl glycine (IVG) which is toxic to the central nervous system (Figure 1) [1]. The pathogenesis of the disease is still not fully understood. Mechanisms thought to be involved include the induction of oxidative stress through accumulating metabolites as seen in the rat brain cortex [2], the reduction of Na+, K+-ATPase activity by free isovaleric acid as shown in synaptic membranes from the cerebral cortex in young rats [3] and abnormal cellular growth signaling through activation of the mammalian target of rapamycin complex 1 (mTORC1), as suggested from studies with human IVD deficient cells [4].
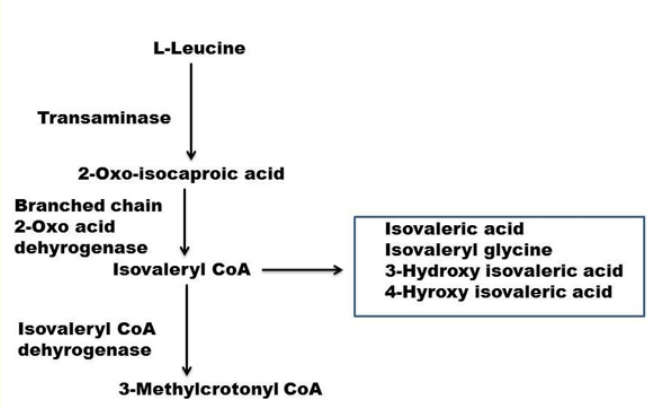
Figure 1: Catabolic pathway of leucine. The names of some of the intermediates are shown. The names of the enzymes are on left. The third step catalyzing enzyme isovaleryl CoA dehydrogenase is deficient. The abnormal metabolites that may be elevated due to the enzyme deficiency are shown on left in the box.
Two clinical phenotypes have been observed in unscreened patients [5]. They may become symptomatic within the first days or weeks of life, presenting with poor feeding or vomiting and severe metabolic acidosis accompanied by neurological signs including lethargy, potentially progressing to coma or death [6]. Alternatively, patients may present later in childhood with acute acidotic episodes often triggered by catabolic stress such as intercurrent illness [7]. Besides, a third distinct phenotype of IVA has been identified by newborn careening (NBS) (1). IVA was first reported by Tanaka., et al. [8] who described the sibling with recurrent episodes of vomiting and lethargy and an unusual odor of “sweaty feet”, in whom a high urinary excretion of isovaleryl glycine and other metabolites of isovaleryl-CoA were detected using gas chromatography (GC) and mass spectrometry (MS).
Patient 1 was delivered after a 38 week-gestation period, normal delivery and discharged 8 days after birth. The infant was well at birth. The patient displayed symptoms such as poor feeding lethargy, hypothermic, tremors and convulsions 12 days after birth. A foul odor “sweaty feet”; was noticed in the female child. Biochemical laboratory examinations indicated elevated levels of ammonia (1,356 μg/dL); therefore, she was subjected to an intravenous administration of benzoate and arginine, as well as continuous hemodiafiltration. Urine isovaleryl glycine (IVG) level found to be 3200 mmol/mol creatinine. Following a diagnosis of IVA, the patient was administered with leucine restricted formula, L-carnitine and L-glycine. The blood ammonia level decreased and was maintained within the normal range; however, severe mental retardation persisted in the patient. Thrombocytopenia, neutropenia and pancytopenia are reported. The patient become cyanotic and lapse into coma followed by death. The cause of death may be severe metabolic acidosis, cerebral edema, hemorrhage or infection. If the patient survives the acute neonatal episode, the subsequent course is that of chronic intermittent form and further development may be normal.
Patient 2 was a female with developed symptoms such as petechiae, poor feeding, vomiting and lethargic within 09 days after birth. Based on the results of a complete blood count, the patient was diagnosed with anemia (Hb 8.2 g/dL) and thrombocytopenia (3,000 per μL). She was subjected to a blood transfusion and treated with antibiotics. Mild mental retardation was identified at the age of 3.5 years. The patient showed symptoms such as repetitive vomiting with mild hyperammonemia (150 μg/dL) at the age of 4.5 years. A diagnosis of IVA was made upon observation of recurrent vomiting with metabolic acidosis (pH 7.3, HCO3 − 9.3 mEq/L) and urine organic acid analysis at the age of 5 years. Urine IVG level found to be 1050 mmol/mol creatinine. The frequency of catabolic episodes was highest during infancy and subsequently decreased because of fewer infections and decreased protein intake which naturally occur with normal growth. Combining early diagnosis with protein restriction and administration of glycine and carnitine has improved the chances of normal development considerably. This case was diagnosed along with other organic acidurias [9].
Blood gas analysis revealed severe metabolic acidosis (pH:7.09) with an elevated anion gap (27.8 mmol/L) and an increased base excess. Hyperglycemia was seen in both the cases. Preliminary urinary screening tests were done. Benedict’s test and Rothera’s tests were positive in the patients.
High-performance liquid-chromatography (HPLC) was used to evaluate the disease-associated metabolite screening. The levels of 3-Hyroxy isovaleric acid and isovaleryl glycine were quantitated in patients’ urine sample. [10] The enzyme deficiency was assayed by detecting 3-Methylcrotonyl-CoA produced in the sample was separated by HPLC and detected using an ultraviolet spectrophotometer [11]. Further the diagnosis was confirmed by molecular genetic analysis. Patient 2 was identified with a missense variant p.G250A in homozygous form which has a minimal effect on hydrogen bonds [12]. The findings were consistent with the milder form of the disease with no severe neurological manifestations. p.R398Q variant in homozygous form was found to be the most deleterious and destabilizing mutation reported in patient 1. This highly pathogenic mutation was previously reported in an Omani patient with IVA [13].
Here the leucine catabolism is affected. Severe metabolic acidosis and neurological deficit are seen. It is often fatal during early childhood. Diagnosis made with a few days of birth were associated with more severe disease and mortality of 33%, whereas children diagnosed later and who had milder symptoms showed lower mortality rate of 3% [6]. The history of consanguineous marriage, sudden infant death and the sweaty feet odor were present in the first patient. Detection of typical metabolites in blood and urine and mutation screening analyses confirmed the diagnosis of IVA [14]. The treatment of acute metabolic decompensation was a high-caloric infusion therapy, correction of the metabolic acidosis and supplementation with L-carnitine and L-glycine [15,16]. Infants who survived this acute episode will go onto have the chronic intermittent form later on in life [17]. A milder form of the chronic intermittent disease also exists. In both forms, acute episodes of metabolic decompensation may occur during a catabolic state such as an infection [18]. In our patients the clinical/biochemical phenotype correlate fairly well with the phenotype predicted by the mutation found. These cases of IVA were identified by newborn screening by Simple biochemical tests along with HPLC analysis.
This cases illustrates that isovaleric aciduria should be kept in mind in the differential diagnosis of hyperglycemia, acidosis and elevated ketone bodies in urine. We suggest HPLC as an easy applicable useful and inexpensive technique for the initial screening of IVA. Urine IVG level may be a better predictor for diagnosis as they correlate well with the severity of the mutation. Molecular genetic analysis will help to differentiate between the acute neonatal presentation and the chronic intermittent form, which have implications in the treatment of the index case.
The samples were collected from Baby Memorial Hospital Calicut, and the analysis was performed at Amrita Institute of Medical Sciences and Research centre, Kochi.
Copyright: © 2019 Narayanan MP. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.