Sachin KR1*, Ramesh H2, Chaya KA3 and Siddartha E S4
1
Junior Resident, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India
2 Professor, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India
3
Assistant Professor, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India
4
Senior Resident, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India
*Corresponding Author: Sachin KR, Junior Resident, Department of Paediatrics, JJM Medical College, Davangere, Karnataka, India.
Received: January 21, 2019; Published: February 18, 2019
Citation: Sachin KR., et al. “Neurocutaneous Syndrome as Failure to Thrive in Neurofibromatosis 1 – A Rare Case Report”. Acta Scientific Paediatrics 2.3 (2019):15-18.
Background: Neurocutaneous syndromes are rare to see as failure to thrive. Neurofibromatosis-1 is autosomal dominant disease due to mutation of NF-1 gene on chromosome 17q11.2 with a prevalence of 1 in 3000. 50% of cases occur due to Germline Mosaicism and 50% of cases as Somatic Mosaicism.
Case Report: 3 year 9 months old, 1st female child born to non-consanguineous married couple presented to CG hospital with complaints of low grade, intermittent fever, non-projectile vomiting 2-3 episodes per day and not gaining weight since 3 months who is previously treated for pulmonary TB under category 1, at 2yr 6 months of age. On examination, multiple (25) café-au-lait spots all over body, where more than 7 were > 5 mm in size, proptosis of both eyes with purely horizontal nystagmus, capillary haemangioma of 2 x 3cm noted on right lower chest, all typical features of severely malnourished- marasmic child were present, all anthropometric parameters were <-3SD according to WHO growth charts. Biochemical analysis shows decreased GH and T3 with normal serological tests and cytology and negative CB-NAAT on gastric lavage. MRI brain shows obstructive hydrocephalus secondary to craniopharyngioma with optic nerve glioma and histopathology shows WHO grade I pilocytic astrocytoma. After stabilising the child with symptomatic treatment, neurosurgeon consultation was sought and advised ventriculoperitoneal shunt placement to relieve hydrocephalus and biopsy to confirm diagnosis. Child withstood the procedure well without any significant neurological deficit. Now child is in follow up at 20 days of post-operative period with good appetite and gaining weight.
Conclusion: In a child presenting with failure to thrive an accurate history, meticulous clinical examination and high degree of suspicion are of paramount importance to reach early diagnosis and initiate appropriate therapy.
Keywords: Neurocutaneous; Neurofibromatosis; Mosaicism; Hydrocephalus; Ventriculoperitoneal Shunt
Neurocutaneous syndromes are rare to see as failure to thrive. Neurofibromatosis-1 or Von Recklinghausen's disease is autosomal dominant disease due to mutation of NF-1 gene on chromosome 17q11.2 with a great variability of expression involving all organs in the body. Prevalence of Neurofibromatosis -1 is about 1 in 3000 [1-3]. 50% of cases occur due to Germline Mosaicism and 50% of cases as somatic mosaicism [1,2]. We are reporting a case, whose mother had developed eruptive neurofibromatosis-1 during her 1st pregnancy [2] due to hormonal imbalance, which is rarely encountered clinically so far.
A 3 year 9 months old, 1st female child born to non-consanguineous married couple with good birth weight, exclusively breast fed for 6 months and good home diet, attained all developmental milestones for her age, completely immunized, presented to tertiary care government hospital with complaints of low grade, intermittent type fever, non-projectile vomiting 2-3 episodes per day and not gaining weight since 3 months. Previously this patient was treated for pulmonary TB (based on history of contact with TB and failure to thrive) who has been given category-1 treatment at 2 years 6 months of her age. On examination, child was found to have multiple (no.25) café-au-lait spots all over body, out of which more than 7 were > 5 mm in size, proptosis of both eyes (left > right) with purely horizontal nystagmus, capillary haemangioma of size 2 cm x 3 cm was noted on right lower chest, all typical features of severely malnourished- marasmic child were present, all anthropometric parameters were < - 3 SD according to WHO growth charts.
On evaluating, the following investigations are revealed
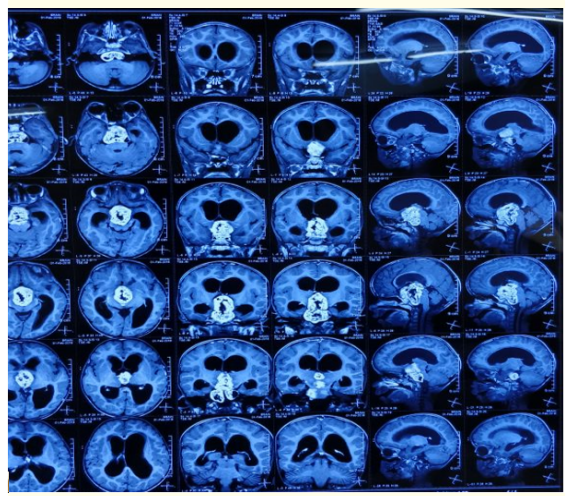
Figure 1
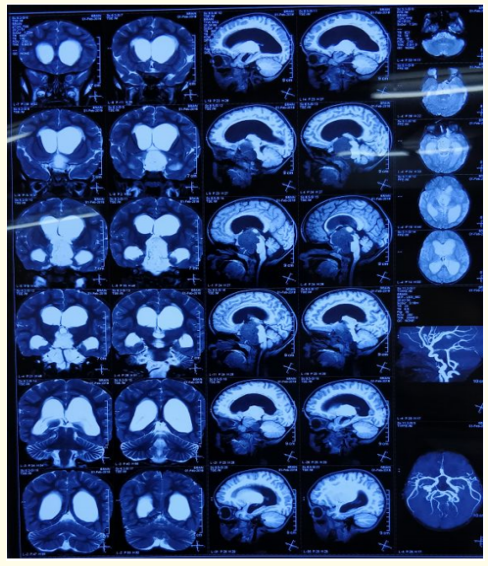
Figure 2
Figure 2: Figure 1 and 2: MRI brain showing obstructive hydrocephalus secondary to well defined lobulated mass in suprasellar
cistern likely to be craniopharyngioma with optic nerve glioma
(left> right).
Histopathology: WHO grade I pilocytic astrocytoma (as shown in figure 3)
After stabilising the child with symptomatic treatment, neurosurgeon consultation was sought and advised ventriculoperitoneal shunt placement to relieve hydrocephalus and biopsy to confirm diagnosis. Child withstood the procedure well without any significant neurological deficit. Now child is in follow up at 20 days of post-operative period with good appetite and gaining weight.

Figure 3: Histopathology examination of the CNS mass shows moderately cellular glial neoplasm composed of bipolar piloid cells with thick processes, which are arranged in perivascular pattern and are dispersed over a microcystic to myxoid glial stroma.
The symptoms of neurofibromatosis have been observed by Robert William Smith in 1849 [4]. The classical variety of symptoms of neurofibromatosis were coined by German pathologist Friedrich Daniel von Recklinghausen in 1882 and hence it is called as von Recklinghausen disease. Neurofibromatosis-1 is inherited as autosomal dominant trait and is caused by dominant loss-of-function mutation in the NF-1 gene. NF-1 gene is located at 17q11.2 and encodes a protein neurofibromin, a tumour suppressor gene. Neurofibromatosis-1 exhibits a wide range of variability of expression and complete penetrance. NF-1 which is the most common type accounts for 90% of cases and is characterized by multiple caféau-lait spots and the incidence of neurofibromas along peripheral nerves [5]. Café-au-lait discoloration generally emerge before the development of neurofibromas and increases when age progresses [6]. Neurofibromatosis type 1 (NF1) represents a major risk factor for development of malignancy, particularly malignant peripheral nerve sheath tumors, optic gliomas, other gliomas, and leukemias, malignancy is an important component of the NF1 phenotype, and one of the few life-threatening complications [7].
Most children with NF1 followed a regular education, over one third of patients needed support, such as language therapy, extracurricular educational support, or psychological support. There is a comorbidity of motor and cognitive deficit in developmental disorders. The principal complication during childhood is learning disabilities [8].
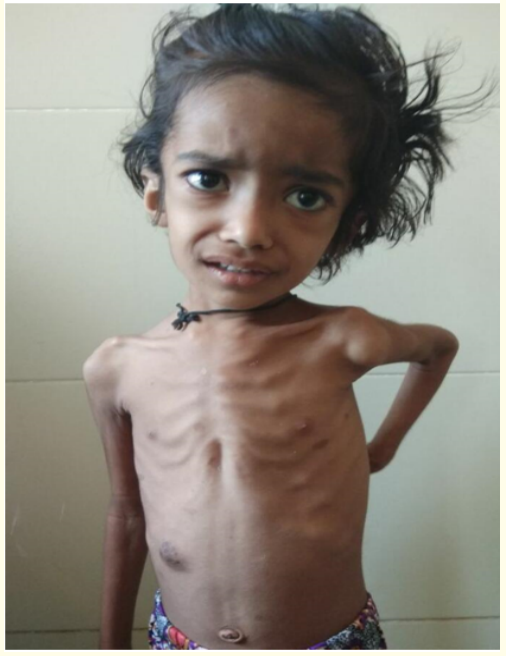
Figure 4: The image shows generalised wasting with prominent ribcage and capillary hemangioma over right lower chest region.
The disease is clinically diagnosed when any 2 of the following 7 features are present [2,3].

Figure 5, 6 and 7: All three images showing café au lait spots more than 5 mm in size.

Figure 8a: The image showing the cutaneous neurofibroma in mother which developed during her 1st pregnancy.

Figure 8b: The image showing café au lait spot over thigh in mother
In our case, about 25 Café-au-lait spots, optic nerve glioma with craniopharyngioma (left > right) and a first degree relative with neurofibromatosis-1 (mother developed cutaneous neurofibromas during her first pregnancy) were present. Management is multidisciplinary follow up which includes yearly ophthalmologic examination, neurological assessment, blood pressure monitoring and scoliosis evaluation. Symptomatic tumours are treated with chemotherapy, radiotherapy or surgical excision.
Neurocutaneous syndromes rarely present as failure to thrive. In a child presenting with failure to thrive an accurate history, meticulous clinical examination and high degree of suspicion are of paramount importance to reach early diagnosis and initiate appropriate therapy. In our case report child fulfils 3 features out of 7 with optic nerve glioma extending into hypothalamus leading to endocrine deficiencies or failure to thrive.
Copyright: © 2019 Sachin KR., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.