Binata Marik1*, Arvind Bagga2, Aditi Sinha2, Pankaj Hari2 and Arundhati Sharma1
1
Laboratory of Cyto-Molecular Genetics, Department of Anatomy, All India Institute of Medical Sciences, New Delhi, India
2
Division of Nephrology, Department of Pediatrics, All India Institute of Medical Sciences, New Delhi, India
*Corresponding Author: Binata Marik, Laboratory of Cyto-Molecular Genetics, Department of Anatomy, All India Institute of Medical Sciences, New Delhi, India.
Received: January 30,2019; Published: February 18, 2019
Citation: Binata Marik. “Whole Exome Sequencing Reveals Novel PHEX Mutations in Patients of Sporadic Hypophosphatemic Rickets”. Acta Scientific Paediatrics 2.3 (2019):12-14.
Hypophosphatemic rickets is a form of refractory rickets that cannot be treated by usual calcium and vitamin D supplementation. It can be inherited or acquired and caused due to mutations in phosphate regulating genes such as PHEX, FGF23, DMP1 and ENPP1 etc. Whole exome sequencing in patients with sporadic hypophosphatemic rickets revealed 3 novel mutations in PHEX gene. This article emphasizes the importance of genetic testing for precise diagnosis, timely initiation of treatment, and better management of the condition, especially in developing countries.
Keywords: Genetic Testing; Genes; Mutations; Phosphate; Refractory Rickets
Hypophosphatemic rickets (HR) is a genetic disorder characterized by hypophosphatemia and osteomalacia. It is caused due to excessive excretion of phosphate in urine due to mutations in genes involved in renal phosphate reabsorption. It can be inherited or acquired. X-linked HR (XLHR) due to inactivating PHEX mutations is the predominant form and an account for approximately 80% of the familial cases [1]. XLHR is inherited in a dominant manner. Autosomal dominant and autosomal recessive forms of HR due to FGF23, DMP1, ENPP1 and SLC24A3 mutations are also documented but their occurrences are rare. Acquired forms include sporadic HR and tumor induced osteomalacia. Clinical features are almost similar in all forms of HR but their treatment modalities are different. Genetic testing can only provide a correct diagnosis. Screening of individual genes is time consuming. Whole exome sequencing (WES) approach allows rapid identification of causative mutations and aids in an accurate diagnosis. We report here the mutations identified in patients with sporadic HR.
Five clinically diagnosed sporadic HR female patients without any family history, born out of non-consanguineous marriage were recruited. Detailed clinical and family history was noted, and 5 ml blood samples collected after taking informed consent. Genomic DNA was isolated and WES was done for all the samples on Illumina HiSeq 4000 sequencer. Data was analyzed using standard bioinformatics approach. Mutations were validated by Sanger sequencing. Crystal structures of the wild-type and the mutant PHEX protein were obtained using the SWISS-MODEL online software (https://swissmodel.expasy.org/). Comparison of the structure of the normal and the mutant PHEX protein was done using the TMalign protein alignment in silico tool (http://zhanglab.ccmb.med. umich.edu/TM-align/).
The patients had short stature, skeletal deformities and hypophosphatemia with mean age of onset at 2.8+1.3 years. WES identified five PHEX mutations in five patients (Table 1) - a known missense (c.1970A>G→Y657C) [2,3], a known nonsense (c.1979G>A→W660*) [3,4], a novel deletion-insertion (c.1337delinsAATAA→F446*) and two novel deletions (c.651_654delACAT→H218Sfs*2, c.665_674delTGGACCAAGC→L 222Qfs*7). The novel deletions and deletion-insertion mutations lead to frameshift which produced premature termination codons respectively resulting in the formation of truncated and non-functional PHEX product (Figure 1). All the mutations identified in the patients were de novo as their parents carried wild type alleles. Variability in disease severity was also observed. The patient having the missense mutation was mildly affected with delayed onset of disease and healing rickets.
This study reports three novel PHEX mutations and suggests that PHEX may be mainly responsible for sporadic hypophosphatemic rickets in India. Genetic testing provides a precise diagnosis of hypophosphatemic rickets, which allows initiation of specific therapy which can minimize skeletal deformities and help in better treatment and management of the condition, especially in developing countries like India.
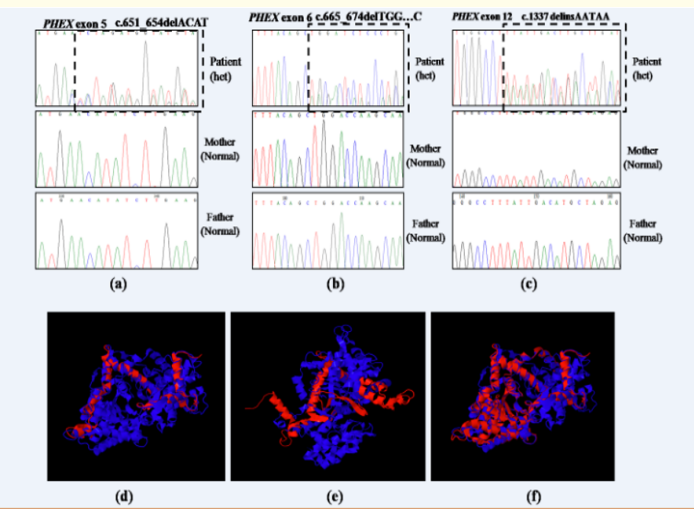
Figure 1: Novel PHEX mutations identified in our study. (a–c) Chromatograms containing novel mutations are shown in comparison to the normal sequences. (a) c.651_654delACAT in exon 5, (b)c.665_674delTGGACCAAGC in exon 6, and (c) c.1337delinsAATAA in exon 12. Superimposed images of normal (blue) and mutant PHEX protein (red). The novel PHEX mutations (d) c.651_654delACAT in exon 5, (e) c.665_674delTGGACCAAGC in exon 6, (f) c.1337delinsAATAA in exon 12 resulted in truncated PHEX protein (red).
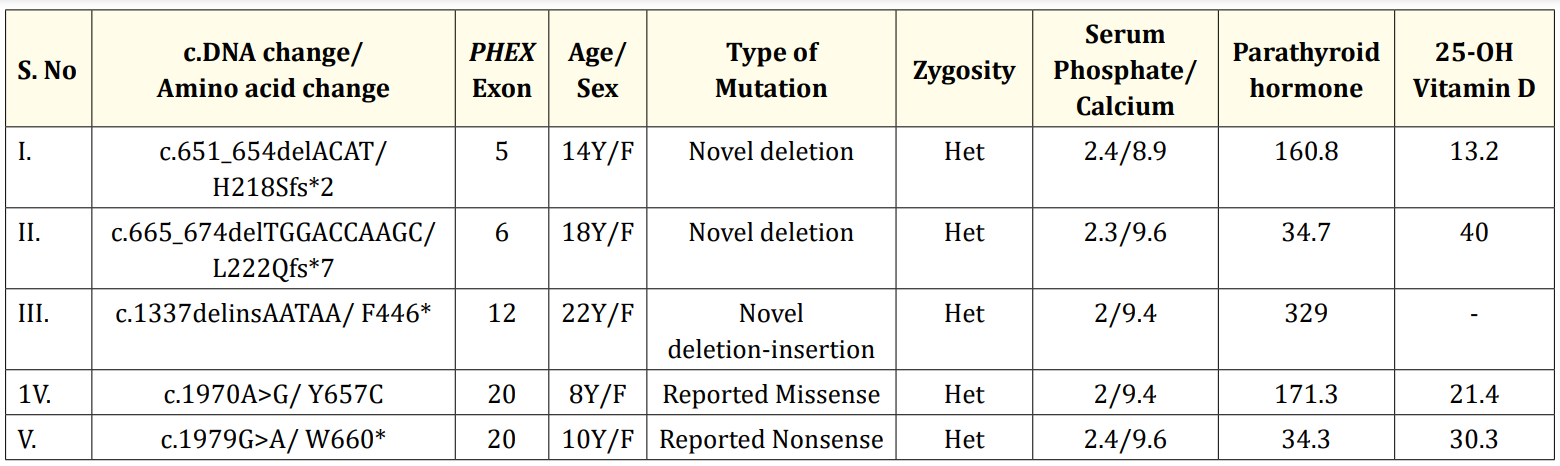
Table 1: Mutations identified in the patients and their clinical characteristics. Het-Heterozygous; Serum phosphate levels -mg/dl, normal range: 3-5mg/dl; Serum calcium levels-mg/dl, normal range-9-11.5 mg/dl; Serum intact parathyroid hormone levels-pg/ml, normal range-15-68pg/ml; Serum 25-OH vitamin D levels-ng/ml, normal range-25-80 ng/ml
None.
We acknowledge the support of the Indian Council of Medical research (ICMR) Project Code: 5/7/946/2013-RCH and Department of Biotechnology (DBT)-India for this study.
Copyright: © 2019 Binata Marik. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.