Gkampeta A1, Kyriaki Papadopoulou-Legbelou2*, Goulordava A2, Keivanidou A1, Thomaidis K3 and Giannopoulos A1
12nd Pediatric Department, Aristotle University of Thessaloniki, AHEPA General Hospital, Greece
24th Department of Pediatrics, General Hospital "Papageorgiou", Aristotle University of Thessaloniki, Thessaloniki, Greece
3Cardiology department, General Hospital of Thessaloniki “George Papanikolaou”, Thessaloniki. Greece
*Corresponding Author: Kyriaki Papadopoulou-Legbelou, Assistant Professor of Pediatrics-Pediatric Cardiology, 4th Department of Pediatrics, General Hospital "Papageorgiou", Aristotle University of Thessaloniki, Thessaloniki, Greece.
Received: August 06, 2018; Published: August 23, 2018
Citation: Kyriaki Papadopoulou-Legbelou., et al. “Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery (ALCAPA) in a Four-Month-Old Infant with Persistent Bronchiolitis”. Acta Scientific Paediatrics 1.2 (2018): 29-32.
Anomalous origin of the left coronary artery from the pulmonary trunk is a very rare congenital heart disease. Without surgical repair, most of these children die during the first months of life. This case report describes a 4-month-old male infant admitted to the hospital as persistent bronchiolitis. Chest x-ray revealed increased cardiothoracic ratio, electrocardiographic abnormalities were indicative for myocardial ischemia and echocardiography along with Magnetic Resonance Imaging unmasked the anomalous origin of the left coronary artery from the pulmonary trunk. As symptoms of congestive heart failure are not specific for this rare anomaly, the origins and course of both coronary arteries should be carefully evaluated in every infant presenting with echocardiographic findings of dilated cardiomyopathy.
Keywords: ALCAPA; Children; Myocardial Infarction
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) comprises a rare congenital heart defect affecting 1 in every 300.000 live births. This anomaly is associated with early infant mortality or adult sudden cardiac death. Infants usually present symptoms of congestive heart failure during the first months of life. After the decrease of pulmonary vascular resistance the anomalous origin of the left coronary artery causes severe myocardial ischemia and left ventricular dysfunction. However, in patients who have developed extensive collateral vessels between the two coronary arteries, the syndrome manifests later in life and even in adulthood causing sudden cardiac arrest [1].
The prognosis depends on early diagnosis and successful cardiac surgery. Without treatment the natural course of the disease is devastating and most children die during the first year of life from heart failure [2]. We report a 4-month-old male infant with signs and symptoms which firstly attributed to persistent bronchiolitis, combined with cardiomegaly, who finally diagnosed with ALCAPA.
A 4-month-old male infant was admitted to the hospital as persistent bronchiolitis (tachypnea, wheezing and reduced feeding for the last two days). He was born full term, with birth weight 3.200g and Apgar score 19 and 510.
On physical examination the infant presented with irritability, fever (38.8°C), tachypnea (respiratory rate 55/min), wheezing, tachycardia (heart rate 190/min), decreased oxygen saturation (SpO2 92%), systolic murmur 1-2/6 at the left sternal border, blood pressure 90/49 mmHg. No other pathological findings in the clinical examination.
Results of laboratory tests, including complete blood count, liver, kidney and thyroid function tests, as well as urinalysis were all within normal limits, except from Troponin T high sensitive (TnThs) and proBNP levels which were both elevated (TnT-hs: 74 pg/ ml - normal ranges < 15 pg/ml, proBNP: 35.000 pg/ml - normal ranges 10 - 51 pg/mL).
Chest X-ray revealed increased cardiothoracic ratio and electrocardiogram (ECG) showed elevated ST segments in leads V1-V4, ST segment depression in leads II, III, aVF, negative T waves and pathologic Q waves in leads I, AVL, V5-V6 (signs of myocardial infarction) (Figure 1). Echocardiography showed left ventricular dilatation (Left Ventricular Internal Dimension in Diastole: 35 mm, normal range: 18 - 25.8 mm) with severe systolic dysfunction (Ejection Fraction 20%), severe mitral insufficiency, dilated right coronary artery (RCA) (Figure 2) and anomalous origin of left coronary artery from the pulmonary trunk (Figure 3). The diagnosis was confirmed by cardiac MRI (Figure 4 and 5).
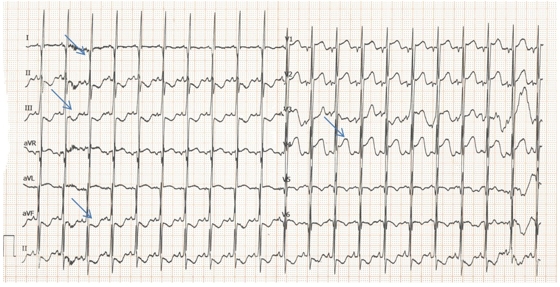
Figure 1: ECG showing elevated ST segments in leads V1-V4, ST segment depression in leads II, III, aVF, negative T waves and pathologic Q waves in leads I, AVL, V5-V6 (arrows).
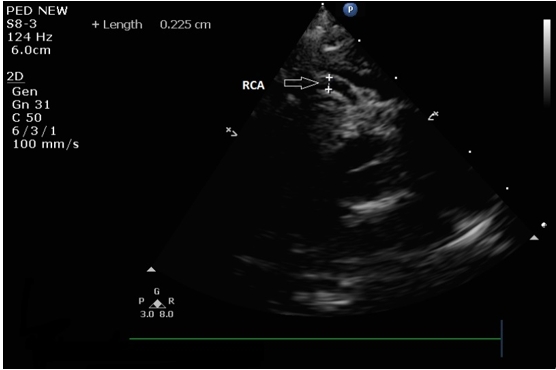
Figure 2: Echocardiogram showing dilated right coronary artery originated normally from the right sinus of Valsava (RCA: Right Coronary Artery).
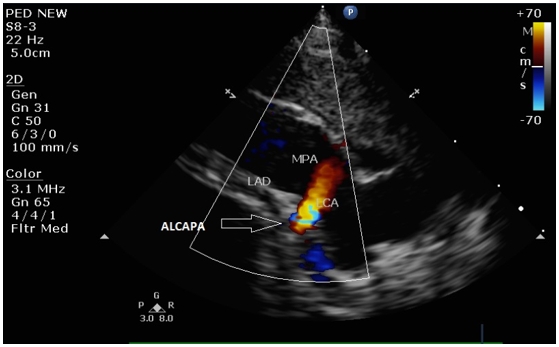
Figure 3: Echocardiogram showing the anomalous left coronary artery from the pulmonary artery. There is retrograde red flow into the main pulmonary artery (LCA: Left Coronary Artery; LAD: Left Anterior Descending; MPA: Main Pulmonary Artery; ALCAPA: Anomalous Left Coronary Artery from Pulmonary Artery).
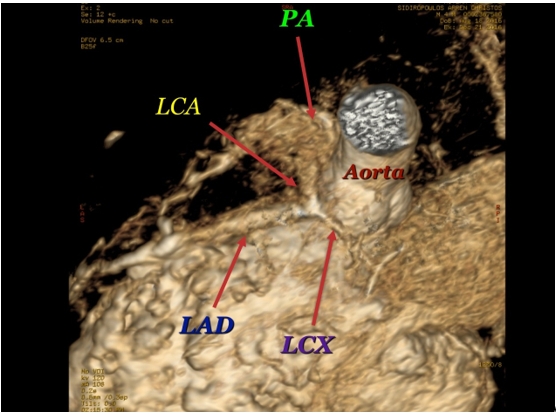
Figure 4: MRI: The left coronary artery (LCA) originates from the pulmonary artery (PA). LAD: Left Anterior Descending, LCX: Left Circumflex.
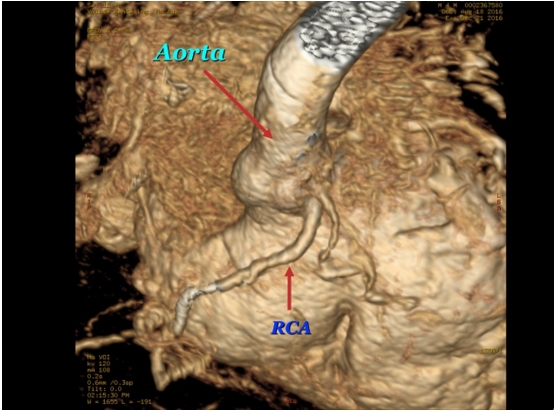
Figure 5: MRI. The right coronary artery (RCA) originates above the right cusp of the aortic valve and the right aortic sinus. At the origin of the RCA is the conus artery.
Immediate cardiac surgery with direct reimplantation of the left coronary artery to the aorta was arranged and successfully performed. The patient's postoperative hospital course was uncomplicated. At 2 months follow-up, the infant remains in good clinical condition with signs of myocardial recovery.
ALCAPA accounts for 0.5% of congenital heart diseases with no gender or race preference. Among untreated cases, the mortality rate within the first year of life is approximately 90%. As for adults, undiagnosed cases lead to death in the fourth decade of life [2].
The low oxygenation in the myocardium of left ventricle, as a consequence of blood flow from pulmonary artery, leads to myocardial ischemia and acute myocardial infarction. Furthermore, the low oxygenation promotes collateral vessels development and right coronary dilatation, while the chronic myocardial ischemia produces ventricular dysfunction and mitral insufficiency [3]. The anomaly is usually isolated, but has occasionally been associated with other congenital heart defects such as patent ductus arteriosus, ventricular septal defect, tetralogy of Fallot, or coarctation of the aorta [4].
The associated clinical picture includes congestive heart failure, cardiomegaly and abnormal ECG pattern. However early diagnosis remains a clinical challenge to the pediatrician because the initial symptoms in infants are nonspecific and older children may not display any symptoms at all. Furthermore clinical manifestations can be misdiagnosed as dilated cardiomyopathy or endocardial fibroelastosis [4,5]. Onset of symptoms is usually observed in the neonatal period (beginning at 1 - 3 months), when feeding or crying induces dyspnea, profuse sweating, pallor and fatigue. Between attacks, physical examination is frequently normal.
In a very recent retrospective study by Patel., et al. the anomalous coronary artery origin was clearly imaged in only 54% of echocardiographic examinations with left ventricular dysfunction being the most common marker in infants [6]. A Chinese study of 27 cases reported acute heart failure (15/27), pneumonia (7/27), and cardiac murmurs with cardiac dilation (5/27) as presenting symptoms [7]. In the study of Guzeltas., et al. 7 out of 12 patients presented with heart murmur and 5 out of 12 patients with shortness of breath [8]. In another very recent study by Gao., et al. including 26 children with ALCAPA aged 45 days to 13 years, the main clinical manifestations were respiratory tract infection, heart failure, dyspnea, feeding intolerance, and failure to thrive [9].
Although few cases of totally asymptomatic adult patients have been reported, a very recent study by Berre., et al. who retrospectively analyzed 11 adult patients with ALCAPA showed that all patients were symptomatic at diagnosis. Symptoms included chest pain (73%), palpitations (64%), heart failure (36%), and/or syncope (9%) [1].
Once the condition is recognized, early surgical option should be offered, as this has been associated with a faster recovery rate [4]. Surgery is recommended in patients with ALCAPA, even in the absence of symptoms, because of the increased risk of ventricular arrhythmias and sudden death. In infancy the prognosis depends on the early diagnosis and surgical treatment [5]. The blood flow restoration in left main coronary artery from the aorta is the primary objective in the surgical correction of ALCAPA. There are several surgical options in the paediatric population, the most popular being direct re-implantation of the LCA into the aorta. The Takeuchi procedure (direct anastomosis of the anomalous left coronary artery to the aorta) has been abandoned in many centers because of a high incidence of main pulmonary artery stenosis [3]. After surgery progressive remodeling and improvement in left ventricular (LV) function may be seen. In cases with very low ejection fraction normalization of LV function usually occurs within 6 months (range 4.5 - 36 months) [9,10].
In conclusion, ECG anomalies (abnormal Q waves and T waves inversion particularly in leads I, AVL, V5-V6) along with LV dilatation and dysfunction in the echocardiogram are indicative for ALCAPA syndrome. The origins and course of both coronary arteries, but especially of the left coronary artery, should be carefully evaluated in every infant presenting with echocardiographic findings of dilated cardiomyopathy.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
The participation involved informed consent.
Copyright: © 2018 Kyriaki Papadopoulou-Legbelou., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
ff
© 2024 Acta Scientific, All rights reserved.