Surgical Management of Hepatobiliary Complications in Sickle Cell Disease. Literature Review
Salah Termos1*, Afaq Mahmoud Alkhalil1, Hassan Al-Jafar2, Ali Alqatan1, Nijmeh Hammoud1, Ahmad Essam Almulla3, Fahad Alabdulghani4 and Mohammad Alhunaidi1
1Department of Surgery, Al-Amiri Hospital, Kuwait
2Department of Hematology, Al-Amiri Hospital Kuwait
3Department of Surgery, Farwaniya Hospital, Kuwait
4Department of Interventional Radiology, Al-Amiri Hospital, Kuwait
*Corresponding Author:Salah Termos, Consultant HPB Surgery, Department of Surgery, Al-Amiri Hospital, Kuwait.
Received:
June 02, 2021; Published: June 23, 2021
Abstract
Sickle cell disease (SCD) is a common hemoglobinopathy that results in vasoocclusive crisis with abnormal endothelial interactions causing repeated ischemia and reperfusion injuries, inflammation and endothelial dysfunction. This pathophysiologic process can affect multiple organs in the body leading to serious medical disorders.
Liver disease is an important factor of morbidity and mortality in patients with SCD. It is most commonly affected due to sickle hepatopathy, an umbrella term for a variety of acute and chronic pathologies that can be manifested in several clinical conditions that range from benign hyperbilirubinemia to an overt liver failure. Management of this disease poses diagnostic and therapeutic dilemmas. Surgical treatment of hepatobiliary complications in SCD patients will be outlined in this review.
Keywords: Sickle Cell Disease (SCD); Hepatobiliary; Complication; Hepatopathy; Liver Failure; Surgery; Liver Transplant
Sickle cell disease (SCD) is an autosomal recessive blood disorder due to genetic mutation and abnormality at the sixth position on the beta-globin chain due to substitution of aminoacid valine to glutamate causing formation of hemoglobin S. This distortion of red blood cell (RBC) causes increased hemolysis and sickling crises. Sickling is a vasoocclusive attacks that lead to microinfarcts in the microvasculature of all tissues and organs resulting in multiple organ failure [1,2].
This Systemic disease can promote a significant liver disease called hepatopathy. Sickle cell hepatopathy is a group of disorders with similar overlapping clinical manifestations lead directly and indirectly to progressive injury to the hepatobiliary system causing decreased liver function up to liver failure [3]. It occurs predominantly in patients with homozygous sickle cell anemia and to a lesser extent in patients with sickle cell trait, HbSC disease and HbSb Thalassemia [4].
In our manuscript, we describe the challenges of liver related conditions in SCD patients and the role of surgery according to different hepatobiliary complications.
Surgical challenges of hepatobiliary complications of SCD
SCD can involve multiple organs and systems including gastrointestinal tract. Hepatobiliary involvement is observed in 10 - 40% cases of sickle cell crises and they can be divided into acute and chronic conditions [8]. This liver dysfunction encompasses a range of hepatic disorders that occur primarily due to sickle disease itself (Hemolysis, sickling consequences, vasoocclusion and lithiasis) or secondarily as a complication of SCD treatment (Iron overload and transfusion problems) [5].
SCD is a complex entity that requires a meticulous clinical care to prevent perioperative sickle disease complications. Incidence of SCD deaths related to surgery is 7% [6]. Attention should be given to hemoglobin level, anesthesia time and complexity of the surgical procedure in order to decrease hypoxia, hypothermia, acidosis and intravascular volume depletion [7].
Surgical management of hepatobiliary complications is usually focused on the etiology and pathogenesis of the disease. In our paper we divided them into biliary and liver conditions.
Biliary conditions
Biliary tract disease is the most frequent indication for surgery in SCD patients. It has no gender predilection. Preoperative optimization of patient’s condition and careful perioperative management can decrease the rate of complications and lead to a better outcome. Procedure complexity or anesthesia time duration increases the risk of acute chest syndrome and other complications [7].
Biliary lithiasis is usually pigment stones that occur due to increased bilirubin production as a result of chronic hemolysis [8]. Gall stones complications including biliary colics, cholecystitis, common bile duct (CBD) stones, gall stone ileus, life threatening cholangitis and pancreatitis in all its forms and manifestations [9]:
- Sickle cholelithiasis: Sickle cholelithiasis (Figure 1) is a common SCD complication. According to Martins., et al. in a study done on 107 patients, the incidence of cholelithiasis in SCD patients was 25%. Among them 50% are symptomatic. Sickle cholelithiasis has an incremental pattern; approximately 70% of sickle patients will develop gallstones at one stage of their life [9,10]. The treatment of symptomatic sickle gallstones is cholecystectomy (Figure 2). Laparoscopic approach is more advocated as it carries less morbidity. In the other hand surgery for asymptomatic patients is still a controversial debate [11].
- Sickle choledocholithiasis: Sickle choledocholithiasis (Figure 3) can be primary formed in the common bile duct or secondary coming from the gall bladder. They are generally treated by endoscopic retrograde pancreatography (ERCP). Failure of ERCP is an indication for surgical common bile duct (CBD) exploration and clearance. Utility of choledochoscope provides direct vision and gives the advantage of stone management in whole biliary tree up to secondary radicals and distal CBD down to the duodenum [12]. In the era of minimal invasive surgery, laparoscopic CBD exploration (Figure 4) is more preferred due to better tolerability and faster recovery. Surgical option to be tailored according to expertise and technical facilities [13].
- Sickle cell cholangiopathy: Sickle cell cholangiopathy occurs as an insult to the bile duct vasculature that leads to ischemia of the biliary system causing sludge and stone formation. Endoscopic sphincterotomy can be beneficial as it may obviate the future development of bile duct stones [14]. Nevertheless, careful evaluation by imaging studies mainly MRCP, prior to any procedure is mandatory as 27% of SCD patients may have jaundice and dilated CBD in the absence of stones [15].
Sequential endoscopic sphincterotomy and CBD clearance (Figure 5) followed by laparoscopic cholecystectomy is a safe and effective approach for the management of cholelithiasis and choledocholithiasis [16].
- Gall stone ileus: This pathology is a rare sequela of cholelithiasis. It occurs as a result of bilioenteric fistula due to erosion by an offending stone. It is manifested by Rigler’s triad (Figure 6), a classic radiologic sign which consists of three elements: 1- Small bowel occlusion, 2- Ectopic gall stones and 3- Pneumobilia [17]. This condition is usually treated surgically according to the condition of the patient with priority being given to relief the small bowel occlusion and biliary obstruction. A literature review generally supports the employment of enterolithotomy in high risk sickle patients and reserving cholecystectomy and division of the fistula for less comorbid patients with feasible anatomy [9].
- Acute pancreatitis: Acute pancreatitis should be considered in the differential diagnosis of abdominal pain in patients with SCD. Pancreatitis is seen in patients with cholelithiasis as a result of common bile duct obstruction but there are also reports of pancreatitis in patients with SCD without cholelithiasis. In these patients, pancreatitis may be due to microvascular occlusion and ischemic injury to the pancreas [18]. Pancreatitis can cause the formation of pseudocyst or walled off necrosis (Figure 7) that can be managed endoscopically or surgically according to size and tissue approximation. Laparoscopic cholecystectomy is advocated in all gallstones related complications. This should be done electively once their condition becomes stable to avoid further attacks.
- Biloma: Common presentation is fever, abdominal pain and jaundice. It occurs as a result of a spontaneous intrahepatic focal biliary stasis. This can rupture and form an extrahepatic intraperitoneal collection [19]. Location of bilomas is detected by different imaging modalities ultrasound, computed tomography (CT), magnetic resonance (MR) and cholangiopancreatography. Gallbladder scintigraphy with technetium-99 may help differentiate biloma from hematoma or a liver abscess [20]. Endoscopic retrograde cholangiography can be a diagnostic and therapeutic option, allowing the decompression of the bile duct and the biliary drainage of the fluid and gas collection [21].
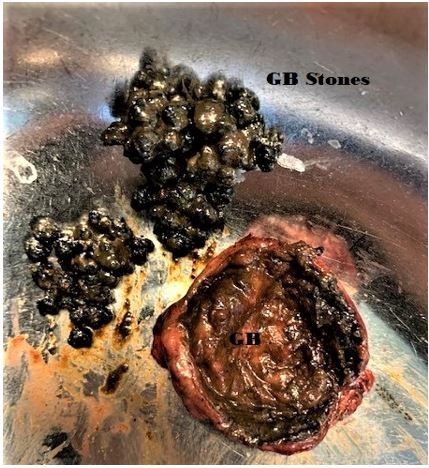
Figure 1: Gallbladder opened showing cholelithiasis (pigment stones).

Figure 2: Laparoscopic cholecystectomy where gallbladder (GB) retracted, cystic duct (CD) and cystic artery (CA) fully dissected.
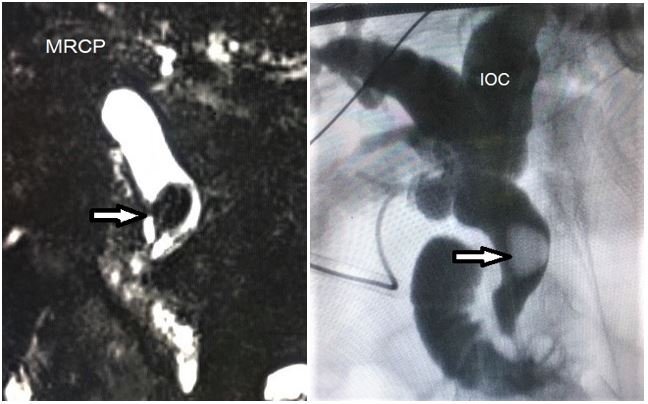
Figure 3: MRCP and intraoperative cholangiogram (IOC) demonstrating choledocholithiasis.
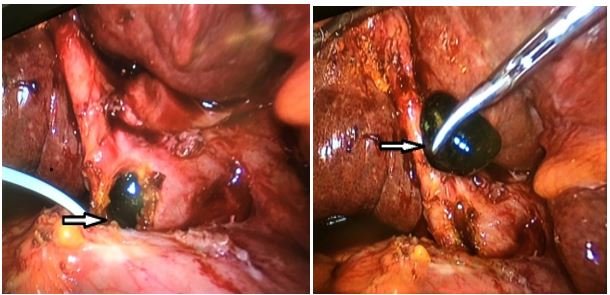
Figure 4: Laparoscopic CBD exploration and stone extraction.
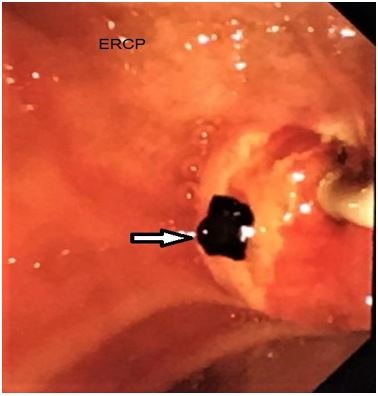
Figure 5: ERCP and CBD clearance (pigment stone extraction).
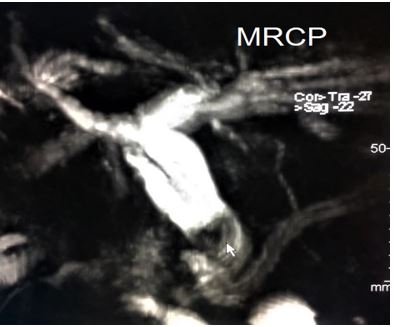
Figure 6: MRCP revealing sickle cell cholangiopathy with CBD stone (arrow).

Figure 7: Enhanced CT scan diagnosing choledochoduodenal fistula manifested with Rigler’s triad (Arrows) [9].
Liver conditions
SCD has evolved into a debilitating disorder with emerging end organ damage. The pathophysiology is a complex mechanism that occurs due to sinusoidal obstruction as caused by sickled RBC, kupffer cell erythrophagocytosis and ischemic induced hyperplasia. Thus, liver injury during hepatic crisis in SCD is quite similar to ischemic hepatitis whose recovery is directly proportional to initiation of proper therapy and alleviation of offending factors [22,23]:
- Sinusoidal hepatic disorders: Sinusoidal hepatic disorders are conditions related to sinusoidal obstruction and red blood cell stagnation. Their consequences are sickle cell hepatic crisis, hepatic sequestration and acute intrahepatic cholestasis. All these disorders if not managed promptly can lead to chronic debilitating liver dysfunction [3,24].
- Transfusion related liver disorders: Transfusion related liver disorders are problems that occur as a result of complication due to repeated blood transfusion in SCD patients:
- Infection: Such as viral hepatitis (HBV and HCV) that if not treated can induce acute and chronic injury to the hepatocyte causing tissue destruction, fibrosis and cirrhosis. This end stage organ damage leads to the formation of dysplastic nodules and cancer transformation to hepatocellular carcinoma (HCC) [25]. Multiple therapeutic options are available as a curative intent or as a bridging therapy before transplant including hepatic resection (Figure 8), local thermal ablation, transarterial chemoembolization (TACE) (Figure 9 and 10), radioembolisation, systemic targeted chemotherapy. However, liver transplantation (LT) is the most effective treatment for both HCC and underlying cirrhosis [26].
- Iron overload: It is also called secondary hemochromatosis. It occurs due to accumulation of iron in the tissues and organs as a result of repeated transfusions due to chronic hemolytic anemia in SCD patients. The most notable organs with iron deposition are the liver, heart, and endocrine glands [27]. The microscopic examination of the affected tissues reveals iron deposition. A liver biopsy can show hemosiderosis as well as fibrosis and cirrhosis [28]. Portal fibrosis can develop in one-third of nonchelated patients within two years, as each unit of packed RBC contains 250 mg of iron. Due to increased risk of liver cancer, annual abdominal ultrasound and alpha-fetoprotein screening is advised [29]. The definitive treatment of decompensated cirrhosis due to secondary hemochromatosis or HCC development is organ transplantation.
- Drug induced liver injury (DILI): Some drugs used in treatment of SCD patients is documented to cause DILI such as Hydroxyurea (stimulate fetal hemoglobin production and decrease number of sickle crisis) and Deferasirox, (chelating agent). Both medications may alter liver enzymes and cause serious complications such as liver failure that might need liver transplant [30].
- Vascular disorders: Vascular disorders are conditions related to anomaly of the hepatic vasculature.
- Outflow disorders: Mainly Sickle Budd Chiari Syndrome, acute or chronic outflow obstruction secondary to hepatic vein thrombosis or fibrosis leading to increased intrahepatic and portal system pressure causing hepatomegaly and ascites then eventually to cirrhosis and liver failure. This condition may require initially transjugular intrahepatic portosystemic shunt (TIPS) (Figure 11) or surgical portosystemic shunting and later LT [31].
- Inflow disorders:
- Arterial etiology: Such as segmental arterial mediolysis (SAM) of the hepatic arteries. SAM is rare non inflammatory vasculopathy due to degeneration of the medial layer of muscular arteries. Most common site is the splanchnic circulation mainly celiac axis [32]. It can be aggravated by SCD and usually manifests with stenosis or aneurysms or thrombosis affecting the arterial blood supply of the liver. This rare condition is generally treated with endovascular interventional radiology or with surgical reconstruction and bypass [33].
- Portal etiology: Due to portal vein thrombosis that can occur in SCD patients due to recurrent crisis and increased VTE risk leading to compromised blood supply to the liver. Extensive thrombosis can lead to serious sequelae up to cirrhosis and liver failure requiring LT [34].
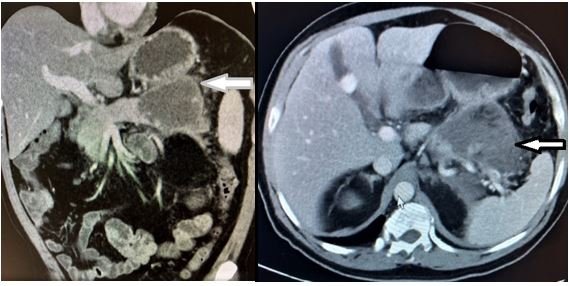
Figure 8: Enhanced CT scan denoting a pancreatic pseudocyst (Arrows).
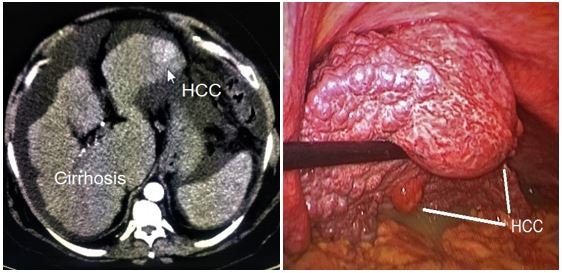
Figure 9: HCC of left lateral segment resected laparoscopically.

Figure 10: TACE procedure of a large HCC in the central part of the liver.
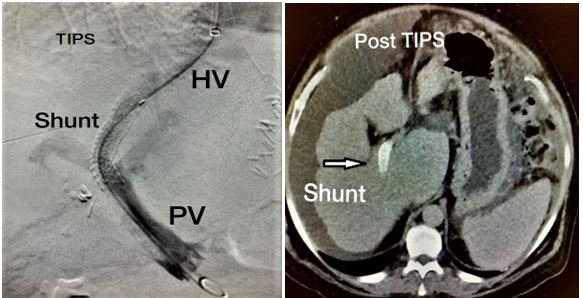
Figure 11: TIPS procedure.
Role of Liver Transplantation (LT) in SCD
Liver dysfunction in SCD is likely to escalade as life span increases and patients incur incremental transfusion overload. Future work must concentrate on not only investigating the underlying pathogenesis, but also identifying in whom and when to intervene with the two treatment modalities available: Exchange blood transfusion (EBT) and LT [35].
Liver transplantation is a complex surgical procedure that removes a diseased liver to replace it with a healthy liver being donated from a cadaveric or living donor. Worldwide experience of LT as a therapeutic modality form end-stage liver disease (ESLD) as a consequence of sickle cell Hepatopathy is slowly developing may be due to the shortage of donors and multiple associated organs involvement in SCD patients.
LT may have a potential role in a selected subset of patients. Two clinical phenotypes that benefit most from LT: First with ESLD without significant sickle related other organ damage and the second perhaps with a milder SCD phenotype and coincident liver disease [36].
SCD and Liver have a reciprocal relation. Both can be triggered and aggravated by each other. Hepatobiliary complications in SCD patients are common and their prevalence increase with age. Surgical management of these disorders can vary from cholecystectomy up to organ transplantation. Accurate indication and proper intervention can ameliorate liver function, improve sickle cell condition and subsequently enhance patient’s quality of life.
Authors declare no conflict of interest.
References
- Satarupa Roy and Subha Ganguly. “Sickle Cell Anemia, an Autosomal Recessive Disorder: A Review”. International Journal of Molecular Genetics2 (2013): 63-67.
- Jensen Frank B. "The dual roles of red blood cells in tissue oxygen delivery: oxygen carriers and regulators of local blood flow". Journal of Experimental Biology21 (2009): 3387-3393.
- Al-Jafar H., et al. “Etiology Based Sickle Cell Disease Hepatopathy”. Open Journal of Gastroenterology 10 (2020): 187-201.
- Galanello R and Origa R. “Beta-thalassemia”. Orphanet Journal of Rare Diseases 5 (2010): 11.
- Sandhya Chandrakar. “Devendra Singh. Sickle cell hepatopathy”. Apollo Medicine4 (2010): 282-285.
- Vinchinsky EP and Lubin BH. “Sickle cell anemia and related haemoglobinopathies”. Pediatric Clinics of North America 27 (1980): 429-447.
- Sarika Ingle and Parvin Ubale. “Anesthetic management of a patient with sickle cell disease for common bile duct exploration”. Journal of Anaesthesiology Clinical Pharmacology4 (2011): 547-549.
- Koskinas J., et al. “Liver involvement in acute vasoocclusive crises of sickle cell disease: prevalence and predisposing factors”. Scandinavian Journal of Gastroenterology 49 (2007): 499-507.
- Jaffar Hussein Asmaa Alrashed., et al. “Gall stone ileus: unfamiliar cause of bowel obstruction. Case report and literature review”. International Journal of Surgery Case Reports 49 (2018): 44-50.
- Raquel Martins Renato soares, The Revista Brasileira de Hematologia e Hemoterapia 39.1 (2017): 28-311.
- Bond LR., et al. “Gall stones in sickle cell disease in the United Kingdom”. British Medical Journal6592 (1987): 234-236.
- Muroni M and Loi V. “Prophylactic laparoscopic cholecystectomy in adult SCD patients with Cholelithiasis: a prospective cohort study”. International Journal of Surgery 22 (2015): 62-66.
- Kelley WE Jr and Sheridan VC. “Laparoscopic choledochoscopy with a small-caliber endoscope. A safe and effective technique for lap CBD exploration”. Surgical Endoscopy 3 (1995): 293-296.
- Dan D and Seetahal S. “Laparoscopic cholecystectomy in SCD patients: does operating time matter?” International Journal of Surgery1 (2009): 70-73.
- Issa H., et al. “Sickle cell cholangiopathy: an endoscopic retrograde cholangiopancreatography evaluation”. World Journal of Gastroenterology42 (2009): 5316-5320.
- Hussein Issa and Ali Al-Haddad. “ERCP in patients with SCD: Diagnostics and Therapeutic Dilemmas”. Gastroenterology Research2 (2010): 74-78.
- Ahmed Al-Salem and Haissam Nourallah. “Sequential endoscopic/laparoscopic management of cholelithisis and choledocholithisis in children who have SCD”. Journal of Pediatric Surgery10 (1997): 1432-1435.
- Narkhede RA., et al. “Laparoscopic management of a proximal jejunal gallstone ileus with patulous ampulla and choledochal cyst”. The Indian Journal of Surgery (2016).
- Ahmed S., et al. “Acute pancreatitis during sickle cell vaso-occlusive painful crisis”. American Journal of Hematology3 (2003): 190-193.
- Lee JH and Suh JI. “A Case of Infected Biloma Due to Spontaneous Intra- hepatic Biliary Rupture”. The Korean Journal of Internal Medicine 22 (2007): 220-224.
- Lebensburge J., et al. “Bi- loma and Pneumobilia in Sickle Cell Disease. Brief Report”. Pediatric Blood Cancer 51 (2008): 153-316.
- Miro AG., et al. “Treatment of Major Iatrogenic Lesions of the Bile Ducts”. Annali Italiani di Chirurgia 73 (2002): 35-39.
- Shah R., et al. “Acute and chronic hepatobiliary manifestations of sickle cell disease: A review”. World Journal of Gastrointestinal Pathophysiology 8 (2017): 108-116.
- Banerjee S., et al. “Sickle cell hepatopathy”. Hepathology 33 (2001): 1021-1028.
- El-Serag HB. “Epidemiology of viral hepatitis and hepatocellular carcinoma”. Gastroenterology 142 (2012): 1264-1273.
- Ali Raza and Gagan K Sood. World Journal of Gastroenterology15 (2014): 4115-4127.
- Powell LW., et al. “Haemochromatosis in the New Millennium”. Journal of Hepatology 32 (2000): 48-62.
- Darbari DS., et al. “Circumstances of Death in Adult Sickle Cell Disease Patients”. American Journal of Hematology 81 (2006): 858-863.
- Porter J and Garbowski M. “Consequences and Management of Iron Over- load in Sickle Cell Disease”. Hematology—American Society of Hematology Education Program (2013).
- Hydroxyurea, Deferasirox Liver Tox: Clinical and Research Information on Drug-Induced Liver Injury”. National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda (2012).
- Aydinli M and Bayraktar Y. “Budd-Chiari Syndrome: Etiology, Pathogenesis and Diagnosis”. World Journal of Gastroenterology 13 (2007): 2693-2696.
- Termos S and Taqi A. “Segmental arterial mediolysis with 5 SAA. A rare fining in a rare disease: Case Report and literative review”. International Journal of Surgery Case Reports 33 (2017): 158-162.
- Serarslan Y., et al. “Nontraumatic spontaneous acute epidural hematoma in a patient with SCD”. Neurocirurgia 25 (2014): 128-131.
- Noubiap JJ., et al. “Sickle cell disease, sickle trait: a systemic review and meta-analysis”. Thrombosis Journal 16 (2018): 27.
- Kate Gardner and Abid Suddle. “How we treat sickle hepathopathy and liver transplatation in adults”. Blood 15 (2014): 2302-2307.
- Hurtova M., et al. “Transplantation for liver failure in patients with SCD: challenging but feasible”. Liver Transplant4 (2011): 381-392.
Citation
Copyright
















 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
