Rodolfo Nunez-Musa*
Contract Research Organization DR (CREOR), MSc, Senior Researcher, Clinical Coordinator at OyMed School of Medicine, Dominican Republic
*Corresponding Author: Rodolfo Nunez-Musa, Contract Research Organization DR (CREOR), MSc, Senior Researcher, Clinical Coordinator at OyMed School of Medicine, Dominican Republic.
Received: October 14, 2024; Published: October 27, 2024
Citation: Rodolfo Nunez-Musa.“Type 2 Diabetes Mellitus and Cardiovascular Disease: Unraveling the Complex Interplay (Part I OF II)" Acta Scientific Nutritional Health 8.11 (2024):45-53.
The relationship between type 2 diabetes mellitus and cardiovascular disease is a complex and multifaceted connection that has been explored in depth by researchers over the years. The risk of developing various cardiovascular complications is significantly higher for those who have type 2 diabetes mellitus, leading to a considerable increase in morbidity and mortality rates. Cardiovascular disease can also have an important impact on the development and progression of diabetes, leading to a vicious cycle that worsens both conditions. Designing effective preventive and therapeutic strategies for both of them requires understanding the underlying mechanisms and shared risk factors between them. With this current paper, we pretend to elucidate the most remarkable intrinsic aspects of this relationship.
Keywords: Diabetes Mellitus; Cardiovascular; Disease; Unraveling; Interplay
Global health challenges of the 21st century are type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD). The prevalence of both diseases continues to rise, placing a significant burden on health systems around the world. T2DM, characterized by insulin resistance and impaired glucose regulation, affects over 400 million all over the world and closely links to lifestyle factors, such as sedentary habits and poor dietary choices. Concurrently, CVD encompasses a group of disorders affecting the heart and blood vessels, including coronary artery disease, myocardial infarction, heart failure, and stroke, accounting for a substantial number of deaths globally.
This paper will explore the complex interaction between T2DM and CVD, highlighting the most relevant underlying pathophysiological mechanisms, the epidemiological trends with the greatest impact, and possible interventions to mitigate the combined effect of these conditions. By delving into the current body of knowledge, we hope to provide clinicians, researchers, and policymakers with a comprehensive understanding of the link between T2DM and CVD, which will ultimately guide the development of personalized approaches to the prevention, management, and care of this type of patient.
Throughout this revision, we will look at the latest research and evidence regarding T2DM and its association with various manifestations of CVD. Our focus will be on how common risk factors, such as obesity, dyslipidemia, and inflammation, can aggravate the intertwined pathology of these diseases. Furthermore, we will briefly explore the potential influence of genetics and epigenetics in mediating the susceptibility to both T2DM and CVD, underlining the need for a holistic approach in tackling these entangled health challenges.
By elucidating the intricate relationship between T2DM and CVD, this paper aims to growing the knowledge on these disorders’ pathophysiology and help seeking the improvement of clinical outcomes, as much as to reduce the burden of disease, and rise the overall quality of life for individuals affected by these conditions. By embarking on this research, we look forward to inspire more research and collaboration to tackle the global impact of T2DM and CVD in an effective manner.
Type 2 diabetes mellitus (T2DM) is a chronic disease with a complex pathophysiology in which the progressive deterioration of the β-cells function in the islets of Langerhans and insulin resistance constitute the breaking point in the total control of the disease. These changes lead to poor glucose tolerance and inappropriately high fasting hepatic glucose production. Its management includes interventions in life habits and in the administration of medications that guarantee safety, effectiveness and adherence, with the fastest and most sustained hypoglycemic response and the fewest possible adverse effects.
The role of oxidative stress (OS) in the development and installation of CVD and T2DM and their complications has gained significant relevance in the definition models of their pathophysiology. In the case of T2DM, excessive OS leads to or favors the appearance of insulin resistance, dyslipidemia, and β-cell dysfunction [1]. Epigenetic alterations, such as changes in DNA methylation, are other factors linked to the pathogenesis of T2DM because they also favor β-cell dysfunction [2,3]. Epigenetic changes do not take place independently, but rather cross and regulate, creating an epigenetic profile that modulates the expression of genes in different cell types, developmental stages, and states of health and disease. Since the estimated inheritance of T2DM is around 10%, epigenetics seems to explain most of the rest of the picture. Environmental risk factors induce epigenetic modifications and with it, the expression of genetic risk factors in T2DM that control the specific intracellular signaling pathways involved in the onset and development of the disease [4]. Although the exact mechanisms underlying the effect of DNA methylation on the pathogenesis of T2DM are still unclear, most studies point to its impact on insulin production, β-cell secretory capacity, and insulin resistance. In the past decades, the management of progressive islet damage has seen a significant increase in attention due to this last aspect, because it is one target of epigenetic alterations, and how this deterioration influences the progression of cardiovascular complications [5].
Cardiovascular events are of particular attention in T2DM, both because of their high incidence and their ability to impair the prognosis and stability of patients, since together with nephropathy; it constitutes a comorbidity with non-reversible systemic repercussions. One of the major goals in the comprehensive management of T2DM is the reduction of atherosclerotic cardiovascular disease (ACVD), which would reduce premature death rates, improve quality of life, and decrease the economic and disease burden of patients. In a 2006 study, the transition to a high-risk category occurred at an earlier age for men and women with diabetes than for those without diabetes, reporting that acute myocardial infarction (AMI), stroke or death from any cause in diabetics of both sexes occurred earlier than in non-diabetics at ages of 47.9 and 54.3 years in men and women, respectively. Therefore, except for diabetics aged 40 years or less, T2DM confers a risk equivalent to aging 15 years [6]. Up to two thirds of diabetic deaths are attributed to CVD, of which about 40% are due to cardiac ischemia, a condition highly related to CVAD [7].
Patients with T2DM tend to have larger atheromatous plaques, bulky atheromas, and smaller coronary arteriolar lumen than those with ACVD without diabetes. This relative increase in plaque volume will be associated with major cardiac events at some point in the evolution of the uncontrolled disease, which plaque appearance can be assessed by the calcium score test, as a potential indicator of this outcome [8]. The characteristic development of atheromatous plaque in diabetics is related to the multiplicity of factors that affect the production, installation, and progress of T2DM, making the comprehensive management still more complex.
Within these factors, and for the purposes of this paper, we are going to analyze insulin resistance, dyslipidemia, hyperglycemia, OS, inflammation and endothelial dysfunction, and review the cardiovascular risk (CVR) within the diabetic clinical approach, all considered crucial in determining the most effective therapeutic strategies for T2DM and for the primary or secondary control of global CVR.
Insulin is a polypeptide hormone, produced and secreted by the β-cell of the islets of Langerhans. Through specific receptors, insulin carries the glucose into the cells, but also intervenes in the control and synthesis of cellular components such as glycogen and triglycerides.
The insulin receptor (INSR) is a tyrosine kinase receptor, encoded by a single gene INSR, which provides instructions for making it. The INSR embeds in the outer membrane surrounding the cell, where insulin binds. The INSR is initially produced as a single long protein that afterwards is cut into four parts: two alpha subunits in the cell surface and two beta subunits inside the cell, all working as a unique receptor. While the insulin binds alpha subunits, the beta subunits trigger signaling pathways to start several cellular functions and each subunit mutually phosphorylate. The activation of different pathways via phosphorylation ultimately leads to the translocation of glucose transporter containing vesicles to the cell membrane, via the activation of SNARE proteins (snap receptors), which primary role is to mediate vesicle fusion with the target membrane, creating membrane-bound compartments. This facilitates the diffusion of glucose into the cell. The protein kinase B (PKB) also involved in the reactions phosphorylates and inhibits glycogen synthase kinase, favoring the process of glycogenesis (storage of glucose), that in due course reduces blood-glucose concentration. When the INSR fail to bind appropriately to insulin, hyperglycemia develops, along with other metabolic disturbances.
The IR explains the failure of muscles, fat, and liver to respond well to insulin and their incapacity to take up glucose from the blood. To compensate, the pancreas increases the insulin production, but cannot achieve positive results and a progressive disorder of lipid and glucose metabolism takes place. The IR usually progresses in silence and manifests clinically years after being present in the form of hyperglycemia, favoring atherogenesis and plaque progression in a curvilinear relationship with vascular disease, even in the absence of hyperglycemia [9]. Sustained elevated glucose levels favor an imbalance between the availability of endothelial nitric oxide (NO) and the accumulation of reactive oxygen species (ROS). This imbalance triggers a chain of pathophysiological events that lead to enzymatic dysfunction in endothelial homeostasis, inflammation and cell proliferation and, more particularly, a strong activation of phosphokinase C (PKC) in the endothelium of diabetic patients in relation to the increased generation of ROS and microvascular OS. These latter events, largely promoted by IR, lead to endothelial dysfunction, potentiating vasoconstriction by means of sensitization of myofilaments to Ca2+ of vascular smooth muscle, and impaired insulin signaling peripherally [10,11]. With greater and more extensive experiences, PKC is a possible therapeutic target for the treatment of vascular complications in diabetics.
The p66Shc protein has a wide participation in mitochondrial and cytoplasmic signaling mechanisms, such as in the regulation of ROS and the induction of apoptosis and life expectancy in mammals. Under conditions of hyperlipidemia, it mediates IR and secretory dysfunction in pancreatic β-cells. In the absence of p66Shc, the lifespan is longer and there is greater resistance to OS and age-related pathologies, providing a better counter action against hypercholesterolemia, ischemia and hyperglycemia [11,12]. In many T2DM cases, gene expression of p66Shc is increased in mononuclear cells in peripheral blood samples and seems to be an important determinant for CVR [13]. The role of p66Shc as redox adaptor protein in IR develops under lipotoxic conditions and with excess body fat. The presence of overweight/obesity augments p66Shc expression levels and causes an impaired ability of exogenous insulin to increase cellular insulin content and secreted C-peptide levels in relation to the lipotoxic condition. Bby means of complex biochemical reactions that include the phosphorylation of the ribosomal protein S6 kinase at Thr389 and of insulin receptor substrate 1 at Ser307, the p66Shc protein mediates the impaired β-cell function and IR by saturated fatty acids and excess body fat [12].
Dyslipidemia accompanies up to 70% of T2DM cases, being a very determining factor in atherogenesis in which low-density lipoprotein (LDL) particles carry the highest specific weight. Lipid alterations in T2DM include elevation of triglycerides (TG), high-density lipoprotein (HDL), and especially high levels of apo B-containing particles. Apo-B is the major apolipoprotein of chylomicrons, very low-density lipoprotein (VLDL), and LDL particles, responsible for transporting lipids to tissues, including cholesterol. High concentrations of apo-B and VLDL are related to a high risk of developing CVD and represent an effective marker of cardiac morbidity, particularly coronary [14,15]. Additionally, high concentrations of apo-B can lead to OS in the endoplasmic reticulum in the liver and IR, which some authors have reported improving with the use of some statins associated with Omega 3 [16].
Dyslipidemia can precede the clinical diagnosis of T2DM by years and can be present in people with normal glucose tolerance and who have shown IR, which seems to suggest a closer association with said resistance than with hyperglycemia [17]. Insulin regulates lipolysis and in situations of IR it rises in the blood, causing hypertriglyceridemia that favors the elevation of VLDL accompanied by a decrease in lipoprotein-lipase activity, so that liver clearance deteriorates due to impaired hepatic uptake of VLDL that leads to lipolipase dysfunction [18].
When T2DM is associated with obesity, the increased persistence of dyslipidemia is potentiated, which favors a greater presence of circulating free fatty acids (FFA), that, in turn, further contribute to impaired insulin signaling and β-cell dysfunction, a factor considered as thrombogenic by some authors [19]. High concentrations of apo-B and VLDL reduce HDL levels since β-cell dysfunction has a negative impact on the regulatory role of HDL which ultimately involves insulin sensitivity and secretion and endothelial protection [20].
Elevated plasma FFA and TG levels, increased apo-B stimulation and VLDL release, decreased HDL levels, and lipoprotein modification are the most likely contributors to the high prevalence of CVD in diabetic patients, which it demands a segmented attention to the problem and a greater degree of stratification of the therapeutic schemes, in a vision of guaranteed pharmacological adherence and affordability.
The risk of death associated with dyslipidemia, when stratified by cholesterol levels, is an element of concern due to its impact on the diabetic population in relation to the non-diabetic population because it increases the mortality rate [21]. Table 1, based on Bertoluci., et al. [22], shows the close relationship between cholesterol levels and the prognosis of T2DM and CVD cases. Cholesterol appears as an independent factor of cardiovascular mortality increased by T2DM and raises the need to innovate effective therapies and adjuvant schemes in the long term and as early as the recognition of the metabolic disorder is achieved, even in the absence of hyperglycemia.
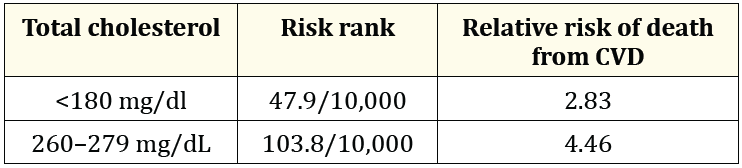
Table 1: Excess absolute risk due to T2DM.
HG plays a seminal role in the development of CVD. High blood glucose values and vigorous antidiabetic therapies have shown a risk for CVD. Its fluctuations elevate the risk of up to 16% for every 1% increase in glycosylated hemoglobin (HbA1c) [23]. HbA1c is the result of the non-enzymatic reaction between glucose and the amino group of the N-terminal valine of the hemoglobin beta chain. The determination of HbA1c represents the measurement of its average percentage in the total population of erythrocytes, which generally reflects the preceding 6 to 8 weeks. Its levels only drop when the erythrocytes carrying HbA1c undergo physiological hemolysis, since substitute erythrocytes are born with normal hemoglobin. The ideal values are located between 5% and 6%. The higher the plasma glucose concentrations, the higher the HbA1c values will be, so they can have a high range of predictability for the development of the microvascular complication and its progression.
From the therapeutic point of view, the presence of elevated HbA1c values is indicative of poor control, so its monitoring and regulation embody a useful tool for the follow-up of the metabolic course in patients with diabetes of any type. However, the finding of increased HbA1c values is not automatically indicative of energetic antidiabetic therapy, since the risks of severe hypoglycemia are more acutely related to fatal cardiovascular events, especially when its decrease is forced to values less than 6% [24].
The American College of Physicians advised against aggressive hypoglycemic therapy for patients with a 6.5% HbA1c level or less and to avoid HbA1c-targeted therapy for patients with a life expectancy of less than 10 years. The American Diabetes Association considers that HbA1c values less than 7% are the therapeutic target and recommends the 6.5% target only if it can be safely achieved, particularly in older populations, as they are more prone to hypoglycemia and more vulnerable to its consequences. Indeed, mean values of 6.9% of HbA1c turn out to be optimal in people aged ≥ 75 years [25,26].
HbA1c favors the intraerythrocytic production of highly reactive free radicals as “advanced glycosylation end products” (AGE) which, by binding to specific cell receptors (RAGE), cause cytokine production and intracellular glyco-oxidative stress, altering membrane functions as well as of various cellular systems. This contributes to endothelial damage by means of hyperaggregation and hyperviscosity that end up inducing inflammation and atherogenic events [27,28]. In addition, the oxidative phenomenon also causes alterations in the metabolism and biodisposition of intracellular iron, culminating in a more unstable compound, ferryl-hemoglobin, which in turn leads to conversion processes into hemoglobin multimers. These multimers promote damage to the vascular intima, which increases endothelial permeability, monocyte adhesion, and macrophage accumulation, thus leading to plaque formation [29]. Therefore, the pathophysiological cascade derived from non-enzymatic protein glycosylation related to chronic hyperglycemia is definitive in the installation of the substrate for the cardiovascular complications of diabetes.
The insufficient counter-regulation resulting from the AGE-RAGE alteration does not allow better antioxidation response and seems to be linked, if not the cause, of the microalbuminuria that accompanies many cases of diabetes [28]. In addition, high AGE concentrations damage vascular smooth muscle, inactivate acetylcholine, and negatively affect NO function, thereby impairing endothelial relaxation and preventing the protective action of NO against plaque formation from the oxidized form of LDL [30]. The longer hyperglycemia lasts, the longer the binding of glucose to hemoglobin and the greater the process of glycation and AGE production. Ultimately, the increased accumulation of AGE in the cell eventually comes out to bind to the RAGEs, and by binding to them, the release of products of inflammation such as cytokines that cause vascular damage initiates. Soluble RAGE markers may have future diagnostic or premonitory value in CVD.
HbA1c is a very reliable predictor of CVD and stroke, fatal or not. An increase of 1 mmol/L (18 mg%) of glucose or one unit in HbA1c increases the risk of macrovascular disease (AMI, stroke or peripheral vascular disease) by 18%. With target values of <7%, the risk of CVD is reduced by 37% at 11 years. Mortality is lower with HbA1c levels between 6% and 6.9%. Fasting glucose in the prediabetes range (100-125 mg/dL) and over the diabetes range (126 mg/dL) moderately and dramatically increases the risk of CVD by 3 to 4 times at 30 years, respectively [31]. The values of HbA1c below 7% are associated with a low risk of macrovascular events and death, and those of 6.5% with a lower risk of microvascular events [32]. However, some controversy remains about the early intensive management of hyperglycemia, departing from the reported stabilization of β-cells function for at least 6 years with short-term initial insulin treatment, followed by intensive insulin or oral hypoglycemic therapy [33]. Other trials have reported the absence of a significant benefit in CVD rates and in overall or cardiovascular mortality with intensive control, but a 2-fold increased risk of severe hypoglycemia and a 47% increase in heart failure [31]. Consequently, the negative effect associated with severe hypoglycemia appears to be a significant counterbalance to the potential benefit of intensive hypoglycemic therapy.
In conclusion, as the microvascular changes that occur in the first stage could be reversible if the patient's hyperglycemia is corrected promptly and that these occur even in the absence of hyperlipidemia, early intervention with the fundamental objective of reducing blood glucose to target levels is mandatory and cannot be postponed. Control strategies should be broad and include not only traditional hypoglycemic agents, but also aim to control or inhibit non-oxidative pathways of glucose metabolism activated by hyperglycemia, especially AGEs, as a mechanism for removing fuel that turn them on.
The OS refers to the intracellular elevation of ROS, which causes damage to lipids, proteins, and DNA. ROS play their physiological role in the biology of the redox system in molecular signaling, which initiates and regulates numerous processes. These processes include activation of the mitogenic-activated protein kinase and extracellular signaling-activated kinase pathways, parts of the signaling system for regulation of normal cell growth that alter gene expression in coordination with superoxide dismutase and brings about cell death. In prokaryotes, there are mechanisms described in which ROS activate transcriptional phenomena for adaptation in stress situations [34]. Therefore, when ROS levels exceed normal limits, causing OS, the resulting alterations have a systemic impact on the organism.
ROS are metabolic by-products derived from aerobic reactions, among which superoxide anion (O2–), hydrogen peroxide (H2O2) and hydroxyls (OH) show inherent properties that give them chemical properties to react on different biological substrates. High concentrations of O2− elevate the requirements of certain amino acids; when H2O2 is raised up, it oxidizes other amino acids and promotes the production of a variety of carboxyls. OH is highly reactive with major macromolecules (RNA, DNA, proteins, and lipids), mostly in the presence of polyunsaturated fats.
Due to the shift at some point of the regulatory mechanisms for the inactivation and degradation of ROS, as well as reactive nitrogen species (RNS), free radicals accumulate, leading an inadequate cell proliferation, accelerated migration, endoplasmic reticulum stress, autophagy, senescence, and necrosis, followed by endothelial dysfunction, further perfusion damage, and accelerated atherosclerosis. Hyperglycemia causes elevation of ROS, as it has been stated, which largely explains the cardiovascular manifestations of T2DM, since ROS and RNS, at normal concentrations, are essential for the signaling pathways that guarantee normal cardiovascular function (Figure 1).
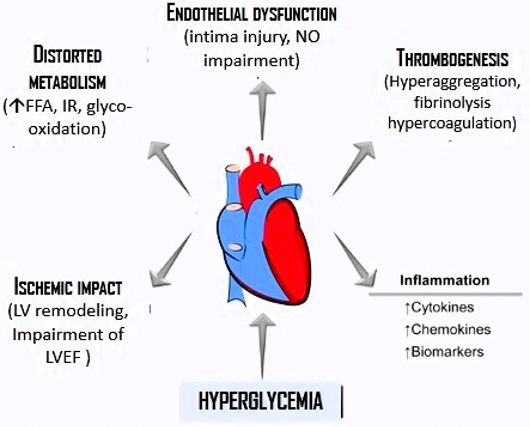
Figure 1: Effects of hyperglycemia on the development and installation of myocardial infraction. Frequently related adverse effects are shown.
At the level of pancreatic β-cells, glycolysis (aerobic or anaerobic) and ROS production are closely linked to their physiological role, glucose sensitivity, and insulin release. The resulting alteration of ROS on the chain of events that determine the function of β-cells, play a fundamental role in the dysfunction and death of these cells [35]. Excessive production or defective removal of these free radicals leads to progressive, sometimes irreversible, damage to cell microstructures through deterioration of their most basic functions at the molecular level, which is essentially OS. In the case of T2DM, the complications associated with OS seem to be related to excess ROS because of a failure in its destruction rate and/or an increase in the production of catalase, superoxide dismutase, and glutathione-peroxidase. As a final balance, the tissues become more susceptible to OS and, consequently, to the complications of diabetes [36], more particularly the vascular ones, which are the main cause of comorbidity and death in T2DM (Figure 1).
OS explains much of the pathogenesis of diabetes, both 1 and 2, where non-enzymatic protein glycosylation, glyco-oxidation and elevated lipid peroxidation cause irreversible damage to enzymes and cellular machinery leading to IR in peripheral tissues, because it affects several points in insulin receptor signal transduction. Its effect is even closely interrelated to the evolution and clinical expression of the disease, depending on the progression and permanence of OS, as demonstrated by Nunez-Musa., et al. [36].
The increasingly better understanding of ultramolecular processes and their macro effects in the organism has allowed us to appreciate a more specific role of OS, such as the regulation of signaling that governs the already mentioned normal or pathological responses at the tissue level. Signaling from the redox system is crucial to define causal management of numerous cellular processes, ranging from mitochondrial functions, as the most appropriate pathway to restore cell differentiation, tissue regeneration, and to prevent senescence. This means extending the therapeutic arm for diabetes to intrinsic causes.
When the balance between the antioxidant and metabolic oxidative systems is broken, the damage described is associated with the elevation of defense products that are useful to estimate the degree of damage caused by OS, called the OS Index (OSI). This products are considered biomarkers to measure OS. Núñez., et al. found highly significant OSI in T2DM and grade II arterial hypertension in a group of patients compared with healthy individuals37. Although more studies and experiences are needed, it is feasible to expect that some specific exogenous antioxidants may have an attenuating effect on the impact caused by OS by inhibiting the initiation or propagation of excess oxidative chain reactions. These antioxidants would act as "scavengers" of free radicals, atomic oxygen quenchers and reducing agents. With the use of precise biomarkers, an opportunity opens up to forecast future events or complications in the broad pathophysiological spectrum of T2DM.
An abnormal inflammatory response may contribute to T2DM by causing hyperglycemia-enhanced IR, which in turn promotes complications. Since the late 1990s, the association of inflammatory mechanisms involving chronic activation of the immune system with an abnormal long-term inflammatory response that does not achieve any tissue repair, as can be found in T2DM [38], has been recognized. In obesity, metabolic syndrome, and coronary artery disease (CAD), inflammatory markers and mediators have been identified, including some useful for the prognosis of events and complications. Subclinical chronic inflammation is a triggering factor in the origin of T2DM. For example, in cases of overweight or morbid obesity, C-reactive protein (CRP) is elevated as a consequence of the abnormal production of tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6) by the adipocytes (Figure 2).
![Figure 2: Cascade of hyperadiposity-related events that explain the underlying chronic and asymptomatic inflammation
linked to the appearance of DMT2 (Based on Stanimirovic [39]).](https://actascientific.com/ASNH/images/ASNH-08-1456_figure2.png)
Figure 2: Cascade of hyperadiposity-related events that explain the underlying chronic and asymptomatic inflammation linked to the appearance of DMT2 (Based on Stanimirovic [39]).
CRP belongs to a conserved protein family called pentraxins, widely identified in several organisms. Native CRP (nCRP) mainly displays anti-inflammatory activities, thus inhibiting the alternative complement activation [39]. When it binds to the cell membrane, nCRP can break down into subunits called monomeric C-reactive protein (mCRP), which are critical molecules that cause and sustain inflammation for long-term. They are also responsible of blood cells and platelets aggregation that end up as a principal contributing factor of complications, like thrombosis when stick permanently within arterial tissue, hence, potentially leading to atherosclerosis and to subsequent acute coronary events. Due to a strong angiogenic effect, they may generate neovascularization of tissues in which are deposited or synthesized [40].
CRP is a common actor in the acute-phase protein activation that performs innate immune functions. It may indirectly induce phagocytosis through activation of complement and complement receptors, or directly through receptors of immunoglobulin G. Phospholipase D, in response to cross-linked IgG, is also activated by CRP without any requirement for further cross-linking. Therefore, CRP is capable of triggering phospholipase D activation and increase phagocytosis [41]. In this way, CRP comprises anti-inflammatory structures by activating the C1q molecule in the classic complement pathway engaging C3 and the terminal-membrane-attack complex, thus leading to pathogen opsonization. Conversely, CRP also acts as a proinflammatory mediator by promoting the release of cytokines [39]. In a revision of Matsumori [42], we find that this chronically activated process conducts to subclinical inflammation, a keystone of IR and consequent development and progression of T2DM, since physiological status never returns to normal baseline. Besides CRP becoming an identifiable marker, the acute-phase protein mediators of the innate immune system, as fibrinogen and serum amyloid A, undergo important biochemical changes far from the expected physiological response to infection or to tissue injury of any kind. Additionally to the high levels of CRP and serum amyloid A, some reports show elevation of sialic acid, haptoglobin and of the plasminogen activator inhibitor and the TNF-α interleukin/cytokine hyperactivation, and low albumin and transferrin levels also takes place, all of them linked with T2DM occurrence, for which this circulating products are predictive markers for the development of T2DM [42]. Hyperglycemia has also been reported associated with neutrophil, d-dimer, and inflammatory biomarkers, and more recently acute-phase reactive and pro-inflammatory cytokines due to COVID-19 in causing inflammation and β-cell damage, particularly in patients with new-onset diabetes43. In all of these circumstances, as subclinical inflammation persists, increased cytokine expressions and immune cell infiltration of pro-inflammatory macrophages can settle in pancreatic islets of patients with T2DM, which explains the important role in defective insulin action and insulin secretion, that eventually will link with fibrosis and amyloid deposits [44].
White adipose tissue (WAT) is part of the overall adipose tissue of the human body and is composed of large adipocytes with a single lipid droplet and few mitochondria, for which it exhibits high-energy storage capacity and homeostasis in response to stressed nutritional demands. It distributes among the viscera and under the skin, and the way in which it distributes, seems to be associated with the development of unfavorable cardio-metabolic conditions. Obesity characterizes by the excessive accumulation of body fat in WAT and by a high production of inflammation molecules, especially TNF-α and IL-6 [45]. Taking into account the frequent and strong T2DM-Obesity-CVAD linkage, this increased production of TNF-α and IL-6 can induce hepatic CRP synthesis and promote cardiovascular complications. Therefore, obesity is a subclinical inflammatory condition that promotes the production of pro-inflammatory factors and that in turn favors the pathogenesis of T2DM and its complications. Hyperlipidemia, within the atherosclerotic plaque, causes the recruitment and migration of monocytes and other immune agents and of the inflammation within the subendothelial layer. These monocytes transforms into macrophages that, once activated, are expressed as capture receptors to facilitate the immersion of native and oxidized LDL. The end-result is the formation of foam cells, which, along with other inflammatory cells, increase the production of chemokines and cytokines [7].
Since these mechanisms always move forward and promote atherosclerotic injury in an inflammatory environment, T-cells from plaque-associated T2DM lesions predominate in the pro-inflammatory Th-17 phenotype. Additionally, in diabetic plaque, cholesterol crystals activate other inflammatory mediators with great impact on the deterioration and severity of the process, such as the neuronal apoptosis inhibitor protein, the leucine-rich repeater protein and the somo complex -inflammatory NALP3 [46]. Much of these phenomena, coupled with abnormal transcription of genes involving NF-kB products and inleukin-1β, may be responsible for the accelerated rate of atherosclerosis in diabetic dyslipidemia, especially in the presence of OS [47].
In particular, the renin-angiotensin system is also involved in inflammation and subsequent damage to the islets. Angiotensin II, regardless of its effects on glucose and its regulatory action on blood presure, promotes inflammation and β-cell dysfunction. These effects ultimately induce the expression of MCP-1 chemokin and IL-6 and, as a consequence, permanent damage of β-cells, mitochondrial function and insulin secretion impairment, together with an increase in β-cell apoptosis [48], hence the trend towards the use of enzyme or angiotensin receptor blockers in patients with hypertension and T2DM and even in cases with risk of developing T2DM [49].
In summary, hyperglycemia and hyperlipidemia promote inflammation by mimicking glucose utilization in conjunction with alterations in oxidative phosphorylation, after which the inflammatory response spreads with cell migration into adipose tissue, islets, and the vasculature. Therefore, glycolipotoxicity is the major contributor to the triggering of the inflammatory response in T2DM that will eventually lead to IR, largely due to oxidative damage to the endoplasmic reticulum and subsequent pathophysiological alterations at the endothelial level. Based on these reasoning, antidiabetic therapy takes into account the anti-inflammatory effects expected in commonly used drugs to reduce blood glucose and control blood lipids. A similar argument applies to anti-inflammatory drugs with a specific molecular effect and with greater impact in blocking the molecular steps of inflammation.
In the second and last part of this review, we will analyze endothelial dysfunction and the interaction of CVR factors in the context of T2DM.
The author declare no conflict of interest.
Copyright: © 2024 Rodolfo Nunez-Musa. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff







