SK Chand Basha* and Mekala Janaki Ramaiah
Department of Biotechnology, Koneru Lakshmaiah Education Foundation, Vaddeswaram, AP, India
*Corresponding Author: SK Chand Basha, Department of Biotechnology, Koneru Lakshmaiah Education Foundation, Vaddeswaram, AP, India. E - Mail: mahashaf@gmail.com
Received: August 22, 2024; Published: September 29, 2024
Citation: SK Chand Basha and Mekala Janaki Ramaiah. “Computational Examination of Microglial TREM2 Structure and its Molecular Docking Analysis against Amyloid-β (Aβ) Ligands with Therapeutic Applications in Alzheimer’s Disease (AD)”. Acta Scientific Neurology 7.10 (2024): 21-52.
Background: Alzheimer’s disease (AD) is a complex and cognitive neurodegenerative disorder effecting millions of people across the globe. Emerging studies suggests that Triggering receptor expressed on myeloid cells 2 (TREM2) associated with late onset Alzheimer’s disease (LOAD). TREM2 are the trans membrane receptors of microglial cells which resides in central nervous system. TREM2 facilitates diverse physiological functions of microglia like phagocytosis. The signalling mechanism of TREM2 is still obscure, pertaining to the same we have proposed a hypothesis in our previous paper. In our hypothesis, we state that the ecto domain of TREM2 exhibit differential binding ability against various forms of Aβ ligands. In the current work we have tested our hypothesis by utilising computational tools.
Objective: Two questions arises from our hypothesis that, whether TREM2 has differential binding affinity with Aβ ligands? And To test whether Aβ oligomer has the highest binding affinity with TREM2 ecto domain? The broad objective of our work is to computationally test the TREM2-Aβ ligand binding ability. The current work may pave the way in identification of potential Aβ ligand which have potential therapeutic applications in AD. To corroborate with TREM2 docking studies, we have conducted similar studies on R47H TREM2, which is a mutational variant of TREM2.
Materials and Methods: I-TASSER webserver and AlphaFold data base were utilised for the structural prediction and analysis of TREM2. Totally, 9 different Aβ ligands were utilised for the study. Rigid docking of TREM2 and R47H TREM2 against Aβ ligands were performed by ClusPro and HawkDock servers to signify the differential docking analysis.
Results: Docking results suggests that Aβ 6 and Aβ oligomer ligands were reported as the potential ligands. ClusPro protein-protein interactions suggests that, for both TREM2 and R47H TREM2, Aβ 6 was the potential ligand with cluster size of 337 and 411 respectively. HawkDock results suggests that Aβ oligomer was the potential ligand for both TREM2 and R47H TREM2 exhibiting docking scores of - 4818.30 and - 4142.15 respectively. I-TASSER and AlphaFold servers predicted models of TREM2 structure.
Conclusion: TREM2 structural analysis done by two different web portals explores further insights. Docking studies of TREM2 and R47H TREM2 suggest that Aβ 6 and Aβ oligomer were the potential ligands having therapeutic potential in AD which requires further experimental studies. Finally, our computational examination suggest that, results were in consensus with the first question of hypothesis and in partial consensus with the second question of hypothesis.
Keywords: TREM2; R47H TREM2; Aβ Ligands; Alzheimer’s Disease (AD)
Alzheimer’s disease (AD), a complex and insidious neurodegenerative disease involving heterogeneous pathophysiological attributes culminating to cognitive dysfunction of the affected individual [1]. As of now, 50 million people across the globe are suffering with dementia, the count may accentuate to 152 million by 2050 [2]. Heterogeneous pathophysiological attributes of AD comprises of amyloid -β (Aβ), Hyperphosphorylated tau, neuronal inflammation, apoptosis and oxidative stress, where in, Aβ is considered as the hallmark pathophysiology of AD [3]. AD falls in two categories (i) Early onset Alzheimer’s disease (EOAD) (ii) Late onset Alzheimer’s disease (LOAD). EOAD is developed due to the mutational variants of the genes - amyloid precursor protein (APP) and the presenilins (PSEN1 and PSEN2) leading to the production of aggregation prone Aβ peptides [4]. LOAD associated with rare mutational variants of microglial immune receptors [5], one of such receptors is Triggering receptor expressed on myeloid cells 2 (TREM2) [6]. TREM2 is a transmembrane signalling receptor, which is expressed in microglial cells of central nervous system (CNS). Native TREM2 structure comprises of three constituents (a) extracellular/extrinsic domain consists of 1-172 amino acid residues (b) transmembrane domain consists of 173-195 amino acids residues and (c) intrinsic/ intracellular domain consists of 196-230 amino acid residues. The transmembrane domain of TREM2 associates with DNAX activation proteins 12 and 10 (DAP12 and DAP10), which are the two adaptor proteins [7]. Aβ binds to the ecto domain of TREM2 in the amino acid range of 31-91 [8]. TREM2/DAP12 complex is essentially required for microglial proliferation, phagocytosis, survival and motility [9], but, the signalling mechanism of TREM2/DAP12 is still obscure [10]. In this backdrop, we have proposed our hypothesis regarding TREM2 signalling pathway in our previous review paper [11] which states that TREM2 receptors of microglial cells along with its DAP adaptor proteins driven signalling cascade facilitates amelioration of Aβ aggregates in AD. In our hypothesis, we hypothesise that TREM2 ecto domain binds to both Aβ monomers and oligomers though with differential efficacy. Due to the high soluble nature of Aβ oligomer, we assume that it may have higher binding affinity with the ecto domain of TREM2, playing an essential role in preventing Aβ fibrillation, in turn obstructs the formation of Aβ plaques. Two questions arises from our hypothesis, that, whether TREM2 has differential binding affinity with Aβ ligands? And To test whether Aβ oligomer has the highest binding affinity with TREM2 ecto domain? The current computational study was designed to ascertain our hypothesis and the computational study of R47H TREM2 was performed for corroboration between WT (wild type) TREM2 and its mutant variant (R47H TREM2) and to ascertain the pathophysiological role of R47H TREM2.
Firstly, we have modelled the TREM2 structure based on I-TASSER and AlphaFold webservers and docking was performed by ClusPro and HawkDock servers. Understanding protein structure is indispensable in construing of various biological processes, despite advances in structural biology experimental techniques, determination of protein structure and functions are tardy and expensive [12]. In the backdrop of this, computational methods emerges as the promising field to predict faster and large scale structural and functional protein models. But, accuracy is often a concern, remarkable progress has been made in computational structure predictions as measured by critical assessment of structure predictions (CASP) estimations [13]. In this regards, Iterative threading assembly refinement (I-TASSER) [14] emerges as credible computational method in protein structure prediction as demonstrated in CASP tests. As the gap between the experimentally determined protein structures and the known sequences of proteins is expanding [15] utilizing artificial intelligence (AI) based techniques for protein structure prediction by the researchers is on the rise [16]. The AlphaFold has been emerging as the most popular AI-based protein structure predictor webserver which was developed by EMBL-European Bio informatics institute (EMBL-EBI) in partnership with DeepMind [17]. As per the CASP14, AlphaFold has been recognized as solution to the problem of protein structure prediction [18].
Computational based protein-protein interactions are crucial in construing underlying cellular and molecular mechanisms of essential biological processes [19]. Based on the function, proteinprotein docking algorithms categorically classified into two major types (i) template free docking and (ii) template based modelling [20], we have employed ClusPro and HawkDock as popular template free docking algorithms. Understanding protein-protein interactions are imperative to explore the insights of complex cellular and molecular processes and organization [21]. Since most of the significant biological interactions occurs in transient complexes, experimental determination of the structures becomes difficult, therefore, computational docking methods emerging as promising and potential tools for prediction of complex interactions. In our study, we have utilized I-TASSER and AlphaFold webservers for protein structural predictions and ClusPro and HawkDock webservers for protein-protein docking. The significance of ClusPro webserver is ranking of the docked models based on highly populated clusters with low energy structures [21]. Similarly, the significance of HawkDock is, it enables to recognize and visualize the key amino acid residues involved in protein-protein interactions. To our knowledge, ours is the first comprehensive computational study of TREM2 structure performed by employing I-TASSER and AlphaFold modellers which were designed by different algorithmic framework. The motivating point to employ two different modellers is to add more confidence in the generated models and to explore novel horizons of structural characteristics. Additionally, structure of R47H TREM2 variant was also predicted by using I-TASSER to understand various structural aspects of TREM2 mutant variant. In line to test our proposed hypothesis, we have conducted docking studies to identify the potential Aβ ligands of TREM2 receptors, for that we have utilized a family of 9 Aβ ligands which were imported from PDB data base. 9 Aβ ligands were: Aβ 40, Aβ 42, Aβ 42A, Aβ 42B, Aβ oligomer, 3 Aβ oligomer types and Aβ 6. In ClusPro, 6 Aβ ligands have been computationally tested; Aβ 40, Aβ 42, Aβ 42A, Aβ 42B, Aβ oligomer and Aβ 6, and 6 ligands in HawkDock; Aβ 40, Aβ oligomer, Aβ 6 and 3 types of Aβ oligomers to identify the best ligand in two different docking algorithms. 3 Aβ ligands; Aβ 40, Aβ oligomer and Aβ 6 were commonly tested in both ClusPro and HawkDock based on its pathological significance. As corroboration studies, similar docking studies were conducted between Aβ ligands and R47H TREM2 variant. The overall objective is to differentiate TREM2 binding affinity with that of R47H mutant variant of TREM2 to predict the phagocytic potentiality of the TREM2 and its R47H variant. The outcome of the study will have pathophysiological and therapeutic implications in AD in the way of identifying the best potential Aβ ligand of TREM2.
Amino acid sequence of TREM2 was imported from Uniprot data base-(https://www.uniprot.org/uniprotkb/Q9NZC2/entry) bearing ID - Q9NZC2 in FASTA format. Iterative Threading ASSEmbly Refinement (ITASSER) web portal (https://zhanggroup.org/ITASSER/) was utilized in 3D structure determination of TREM2. After uploading the amino acid sequence the results were generated with job ID S738304. Significantly, I-TASSER generates top 5 predicted models of TREM2 structure. Top Model of TREM2 (Model 1) comprises of 3 structural parameters: C -Score; Estimated TMScore and Estimated RMSD. The rest of the top models (2-5) comprises of C-Score parameter. In addition to it, other parameters of the structure predicted also generated; Top 10 identified structural analogs in the data base of PDB, ligand binding sites, enzyme commission (EC) numbers and active sites.
Amino acid sequence of R47H TREM2 variant was imported from PDB data base-(https://www.rcsb.org/structure/5UD8) bearing ID - 5UD8 in FASTA format and the results were generated with ID S744094. Prominently, I-TASSER generates top 5 predicted models of TREM2 structure. Top Model of TREM2 (Model 1) comprises of 3 structural parameters: C -Score; Estimated TM-Score and Estimated RMSD. The rest of the top models (2-5) comprises of C-Score parameter. In addition to it, other parameters of the structure predicted also generated which comprises of top 10 identified structural analogs in the data base of PDB, ligand binding sites, enzyme commission (EC) numbers and active sites.
I-TASSER web server: Iterative Threading ASSEmbly Refinement (ITASSER), is a hierarchical protocol designed for automated prediction of protein structure and structural dependent function annotation [12]. I-TASSER server was first established in 2008 [22]. Since then it generates numerous protein structure predictions, and the algorithm which were programmed for I-TASSER were tested extensively for its efficacy in both blind tests [23-26] and benchmark studies [27,28]. The protocol of I-TASSER awarded with top server rank for automated prediction of protein structures at 7th to 11th CASP competitions [25]. The work flow of I-TASSER server comprises of 3 steps (i) identification of structural template (ii) iterative structure assembly and (iii) structure based function annotation (Figure 1). In the first step by using LOMETS algorithm, I-TASSER identifies homologous structural templates from the PDB library. LOMETS, which is a meta threading algorithm comprises of multiple individual programs of threading. The continuous aligned fragment structures extracted from LOMETS were reassembled to construct the full length models. SPICKER algorithm utilizes Monte Carlo simulation trajectories to identify lowest free energy confirmation. To refine the modelled structures full atomic simulations which utilizes FG-MD [28] and Mod Refiner were deployed. BioLip was utilized to match its proteins with the I-TASSER models to decipher the physiological functions of the target proteins. To run the I-TASSER, the amino acid sequence of the desired/target protein is required to predict its structure as well as biological functions. I-TASSER web server uses psspred algorithm to predict the secondary structure of the target proteins. The psspred algorithm functions by untangling PSI-BLAST profile data and 7 neural network predictors from varied parameters. SOLVE program was utilized to predict the solvent accessibility of the target protein and ResQ for prediction of normalized B-factor [12]. During the threading alignment process by LOMETS, I-TASSER only utilizes the highest significant templates, where in, the significance was measured by the Z-score. Z-score is calculated in terms of standard deviation, in the way of variation in its raw and average scores. LOMETS generates best 10 templates and utilised in the replica exchange monte carlo simulations as the beginner models in low-temperature replicas. I-TASSER generates top 5 predicted structural models of the target protein. The important parameters in the returns of top models were C- score, TM-score and RMSD, with C-score is utilised in ranking the models. [29].
C-Score: Confidence score estimates the confidence of each generated structural model, its normal range is [-5, 2] and is defined by the equation as follows
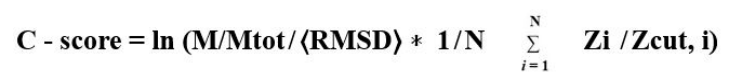
M/Mtot-Denotes the ratio between number of decoy structures in SPICKER cluster and the total decoys produced during I-TASSER simulations.
⟨RMSD⟩ - Average RMSD of decoy structures to the centroid of the clusters.
Zi ̸ Zcut, I-Normalized Z-score of the top template gene measured by LOMETS.
TM-Score and RMSD: Template modelling score (TM-score), measures the structural similarity with normal values falling in the range of [0, 1] [30]. Root mean square deviation (RMSD) estimates the average distance between the atoms of proteins which were super imposed. Generally, RMSD values of less than 1Ao is appropriate.
COACH algorithm generates the ligand binding site predictions of target models, whereas Gene ontology (GO) term prediction and Enzyme commission (EC) number were generated by CO-FACTOR algorithm.
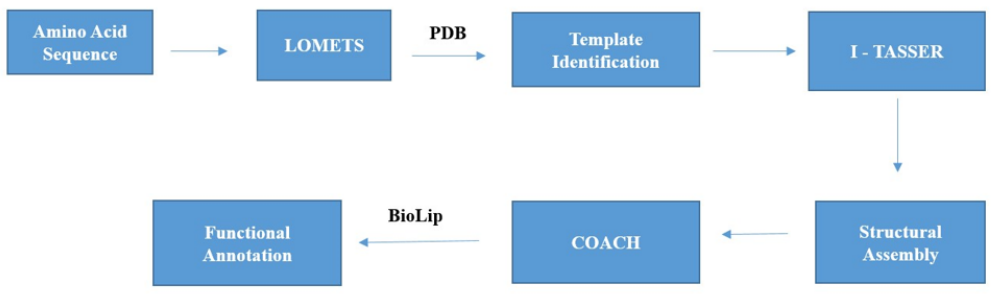
Figure 1: Schematic representation of I-TASSER Protocol.
The 3D structure prediction of TREM2 protein was generated in the AlphaFold database (https://alphafold.ebi.ac.uk/entry/Q9NZC2) and was utilised in our study for analysis of TREM2 structure. In AlphaFold, the predicted structure of protein comprises of 3 outputs besides basic information drawn from Uniprot and PDB they are (i) 3D atomic co-ordinates (ii) pLDDT (local distance difference test), which estimates the per residue confidence in the range of 0 to 100, with greater the score corresponds to greater confidence (iii) predicted aligned error (PAE), which is a prediction of pairwise confidence.
The AlphaFold database of protein structures is an extensive and open accessible data base of higher accuracy prediction of protein structures [17]. AlphaFold is an artificial intelligence (AI) based web server developed by EMBL-European Bioinformatics institute (EMBL-EBI) in partnership with Deep Mind [16], which unequivocally expands the basket of protein structure with higher accuracy in prediction. The emergence of AlphaFold in the field of bioinformatics is of utmost importance, as the gap between the experimentally determined protein structures and known protein sequences is huge, which severely limits our understanding of protein structure. Recognizing its accuracy in prediction of protein structures, CASP14 (critical assessment of protein structure prediction) in 2020 designates AlphaFold as a solution to the problem of protein structure prediction [31]. Despite the greater advancements, the current software program attained remotest atomic accuracy in protein structure prediction, particularly when no homologous structure is available [32].
By integrating novel architecture of neural network and training process which relies on geometric, physical and evolutionary constraints of proteins AlphaFold greatly increases the prediction rate of structures [16]. In particular, AlphaFold server incorporates Novel features for instance, embedding pairwise features and multiple sequence alignments (MSA), novel equivariant attention neural architecture, and utilizing self-estimates of accuracy and self-distillation to learn from unlabelled sequences of proteins [16]. Most importantly, Evoformer, which is a significant building block of neural network has been employed to view the predicted structures in the way of a graphical inference problem in 3D space. All such novel architectural neural features greatly enhances the accuracy of protein structure prediction.
In AlphaFold, the predicted structure of proteins comprises 3 outputs besides basic information drawn from Uniprot and PDB, which comprising of (i) 3D atomic co-ordinates (ii) pLDDT (local distance difference test), which estimates the per residue confidence in the range of 0 to 100, with greater the score corresponds to greater confidence. pLDDT objective is to analyse the local accuracy of the predicted structure [33]. Different confidence scores are generated based on the pLDDT scores as follows. (a) Very high model confidence: ≥ 90 (b) Confident: 90 > pLDDT ≥ 70 (c) Low confidence: 70 > pLDDT ≥ 50 (d) Very low confidence: pLDDT < 50 [34]. Very low confidence scores of pLDDT corroborates with higher propensities for intrinsic disorder [35]. The third significant output of AlphaFold is predicted aligned error (PAE), which is a prediction of pairwise confidence [17]. PAE aids in assessment of the relative domain position’s reliability and its orientation, in addition to global topology of the protein structure. Pairwise PAE values are used in colouring the plot, with dark green depicted as higher confidence. The atomic co-ordinates in AlphaFold server are available in PDB and mmCIF formats, whereas PAEs are available in JSON formats [17].
Aβ Ligands used for docking against TREM2: Altogether, 9 Aβ Ligands were utilised for the docking studies against TREM2, and R47H TREM2, the coordinates of the same were imported form the PDB data base (https://www.rcsb.org/). The PDB IDs of the respective Aβ ligands comprises: Aβ 40-1AML; Aβ 6-2ONV; Aβ 422MXU; Aβ 42 A-8EZD; Aβ 42B-8EZE; Aβ oligomer-7ROJ; Aβ oligomer types-3Q9I; 3Q9H and 3Q9J respectively. ClusPro (https:// cluspro.bu.edu) and HawkDock servers (http://cadd.zju.edu.cn/ hawkdock/) were employed for the docking studies. In Both ClusPro and HawkDock servers, both the receptor (TREM2 and R47H TREM2) and ligands (Aβ Ligands) were submitted as inputs in PDB format.
Aβ plaques are the chief pathophysiological characteristic of Alzheimer’s disease (AD), the same is the aggregated product of Aβ monomers in a cascading manner. Initially, Aβ monomers aggregates to form oligomers, which forms protofibrils, in turn aggregates into Aβ fibrils, which assembles to amyloid plaques [36]. Therefore, keeping in view of the pathophysiological characteristic of Aβ monomers and oligomers, we have tested the binding affinity of a family of Aβ monomers; Aβ - 6; Aβ-40; Aβ - 42; Aβ - 42A and Aβ - 42B (number corresponds to amino acid residues) and Aβ oligomer and its types against TREM2 and R47H TREM2 mutant variant. The finding will enable us to understand the differential ligand binding ability of TREM2 and R47H TREM2 and potential ligand of TREM2 which have therapeutic implications in AD.
Aβ - 40 (A4-(1-40)) is one of the major monomeric peptide which cumulatively aggregates to form deposits of amyloid plaque in Alzheimer’s disease. Aβ - 40 peptide was synthetically prepared and its structure is determined by 2D NMR spectroscopy and restrained calculations of molecular dynamics [37]. Aβ - 42 (Aβ (1- 42)), is one of the chief monomeric pathophysiological characteristic in AD, the propagation of the misfolded aggregates of the same provokes AD cascade. The structure of the Aβ - 42 is determined by solid state NMR [38]. Aβ - 42 is known for its polymorphic characteristic, Lee., et al. [39] reported two forms of Aβ-42 structures - Type A and type B (Aβ - 42 A and B) fibrilar structures, which are derived from Alzheimer brain tissues and are determined by cryo-EM. Gray., et al. [40] determined the structure of Aβ-oligomer (asymmetric dodecamer) by IMS-MS, derived from the brain slices of the mouse model KV11. Liu., et al. [41] studied the crystal structures of 4 Aβ oligomeric polymorphic forms (tetramers), based on the macrocyclic peptide model system. Aβ - 6 is a peptide of 6 amino acid residues - GGVVIA which forms amyloid fibril and is derived from amyloid-β (Aβ 37-42) of the Alzheimer’s disease [42].
Protein-Protein interactions are essential for construing of cellular organizational and functional domains [21]. Generally, X-ray crystallography is deployed for mechanistic interpretation of protein-protein interactions at atomic level. But, significant biological interactions takes place in transient complexes, which is difficult to determine by employing empirical structure determining techniques. Based on this background, computational docking methods have been designed and developed to determine the structure of protein-protein complexes, trying to attain accuracy near to X-ray crystallography [43]. There are two types of docking methods: direct and template, direct methods are based on the principle of thermodynamics, which tries to elucidate the structure of target complex accompanying conformational space with low (or) minimum Gibbs free energy. To moderate this complex, feasible computational free energy evaluation model and potential minimization algorithms are required [44]. The important feature of direct docking method is it will generate good results, if the conformational changes of protein-protein associations are moderate [21]. Template docking relies on the aspect that interacting pairs shares more than 30% sequence similarity paving the way to homology based prediction of targeted complex structure, if the related template complex of existing/known structure is accessible [45].
ClusPro webserver was introduced in 2004 [46]. ClusPro server conducts docking operation in three computational steps, which comprises of (1) Rigid body docking: Sample of billions of confirmations; (2) Root mean-square deviation (RMSD) dependent clustering of 1000 lowest energy structures produced to identify the largest clusters which will better represent most probable models of the complex; (3) Energy minimization based refinement of selected structures [21]. PIPER program based on Fast Fourier Transform (FFT) has been utilized in the rigid body docking [47]. In this approach, receptor is placed on a fixed grid, and the ligand is placed on a mobile grid, where in, the interaction energy is represented in the way of correlation function. The crux point in the success of rigid body approach is that the shape complementarity permits certain overlaps, and therefore, this approach could able to resist moderate differences between unbound and bound structures. One of the distinct feature of ClusPro is selection of highly populated clusters of the lower energy structures.
The size of individual cluster depicts the width of respective energy well. The generated docking calculations will be performed in 4 different energy parameter sets viz….Balanced set (by default), electrostatic favoured, Van der waals + electrostatics and hydrophobic favoured [21]. The ranking of the models is based on the size of the clusters, instead of energy, it is based on the aspect that energy estimated by PIPER doesn’t directly associated with binding affinity [48].
The HawkDock is a rigid body and template free integrated server [49], which combines ATTRACT docking algorithm, HawkRank program [50], MM/GBSA and 3D mol.js [51]. Initially, ATTRACT [52], which is a randomized global search program performs the rigid body docking to predict protein-protein complex of two unbound structures, where in, distance squared cut off and maximum steps of the minimization are configured at 50.0 A2 and 1000 respectively. ATTRACT algorithms generates the best 10000 decoys, which are rescored by HawkRank program. Consecutively, 1000 best decoys generated each by ATTRACT and HawkRank are clustered based on the clustering method of Fraction of common contacts (FCC), the thresh hold of the same is set at 0.5.
In each cluster, the best scored model is extracted and are re ranked by HawkRank scoring as top ranked structures. In addition to it, molecular mechanics/generalized born surface area (MM/GBSA), which is an analysis platform of free energy decomposition is utilized. This toll assess the key amino acid residues in the protein-protein binding interface, followed by re-ranking of the top 10 docking models. Finally, 3Dmol.js, which is a web GL-based molecular viewer platform is utilized for interactive exhibition of top models.
The HawkDock server works on python framework of Tornado [49], after submitting the jobs, the results will be generated in the way of seven components (i) input files comprising of receptor and ligand PDB files (ii) text file docking scores of top 100 models (iii) top 10 and 100 models represented in the way of compressed tar files (Figure 2).
Pymol software (4.60 version) was used for the visualization of our computational study.
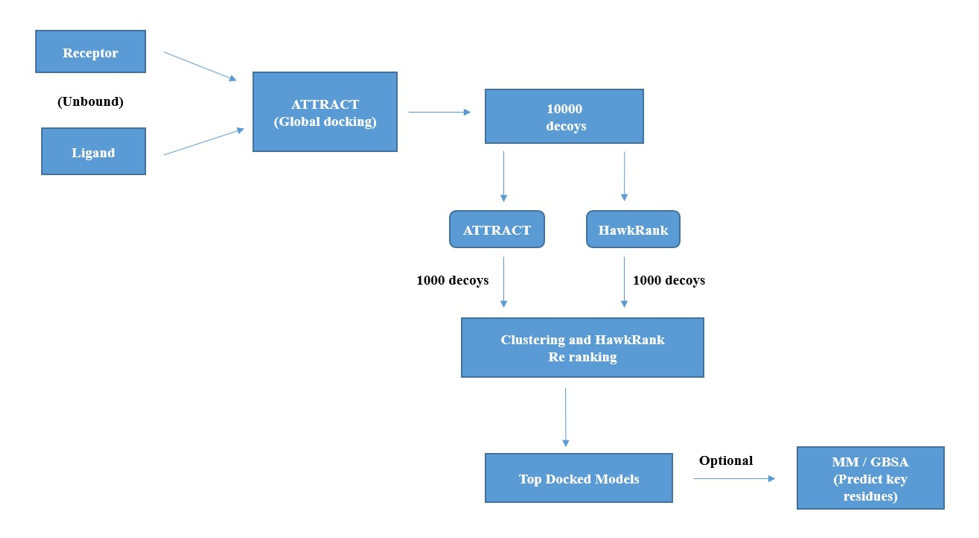
Figure 2: Schematic representation of workflow of HawkDock Docking (Unbound receptor - ligand).
I - TASSER web server generates decoys of TREM2, which are the large ensemble of structural confirmation. SPICKER program of I-TASSER clusters the TREM2 decoys by relying upon similarity of pairwise structure. With respect to the five largest clusters of the structures, 5 top models of TREM2 were generated Figure 3 (a-e). C- score measures the confidence of each TREM2 model. C - score is measured based on the significance of alignments of threading template and convergence parameters of simulations of structure assembly.
Typical range of C - score is [- 5, 2], higher the C - score value, greater the confidence of the generated model. TM score and RMSD values are measured based on the C - score and length of the protein. The top 5 models of TREM2 are ranked based on the cluster size. The top model of TREM2 have a C - score of -5, and its estimated TM score was 0.19 ± 0.04. Estimated RMSD score was 20.4 ± 1.6 Ao. C - score of other models were represented in (Figure 4). The best 10 threading templates of TREM2 utilized by I-TASSER represented in (Figure 5). Other results were also generated by I - TASSER which comprises of predicted Secondary structure of TREM2 (Figure 6 (a-b); predicted Solvent accessibility of TREM2 (Figure 7 (a-b); top 10 Identified structural analogs of TREM2 in PDB (Table 1); ligand binding site of TREM2 (Table 2) and enzyme commission number and active sites (Table 3).
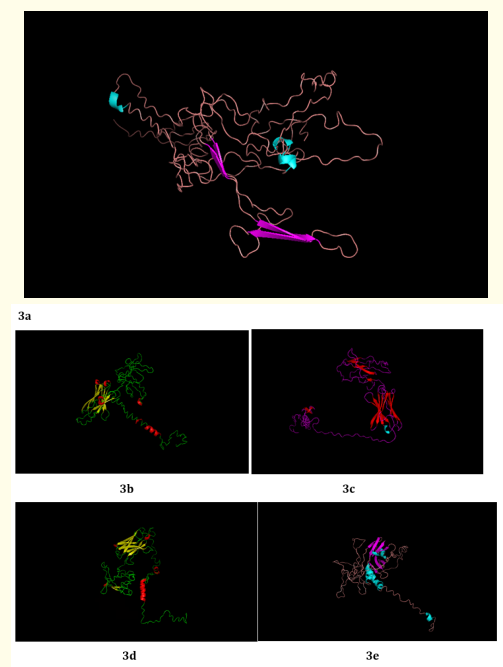
Figure 3: (a - e): Top Model (Model 1), 2, 3, 4 and 5 of TREM2 3D structure prediction by I-TASSER.

Figure 4: C- scores of TREM2 Models (1-5).
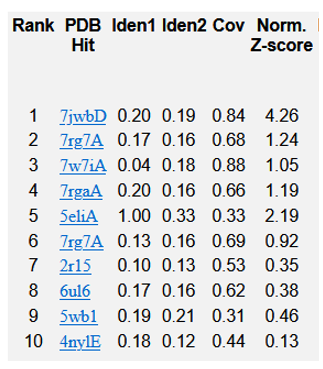
Figure 5: Top 10 templates of threading utilised by I-TASSER for TREM2.

Figure 6: (a-b): Predicted Secondary structure of TREM2.

Figure 7: (a-b): Predicted Solvent accessibility of TREM2.
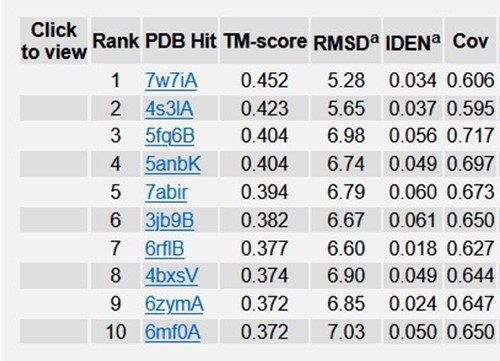
Table 1: Top 10 Identified structural analogs of TREM2 in PDB.
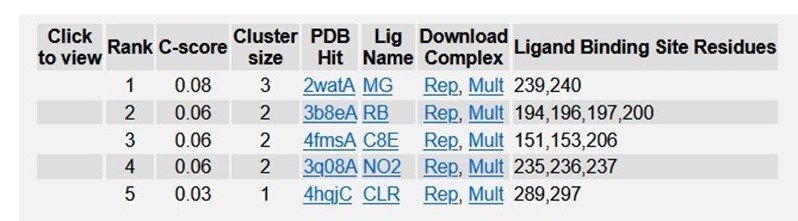
Table 2: Ligand Binding sites of TREM2.
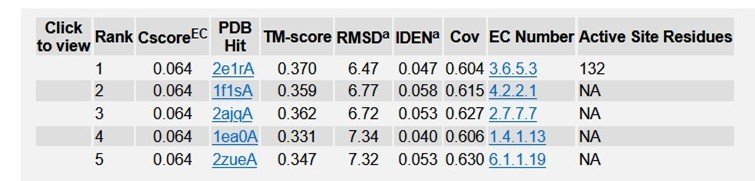
Table 3: Enzyme Commission (EC) numbers and active sites of TREM2.
AlphaFold generates 3D structure of TREM2 (Figure 8) with major outputs i) predicted local distance difference test (pLDDT) (Figure 9) and ii) predicted aligned error (PAE) (Figure 10). pLDDT measures whether the distance of predicted residue akin to its neighbouring C - α atoms. As such following amino acid residues designated under very high confidence range (pLDDT > 90): 21 - 53; 60 - 130; 184; 187 - 190; 192 - 193. Similarly, amino acid residues 5, 6, 9, 20, 54 - 59, 131 - 133, 175 - 183, 185 - 186, 191 and 194 - 198 falls in high confident category (90 > pLDDT >70). TREM2 amino acid range 1-4, 7, 8, 10 - 19, 134 - 143, 169, 171-174, 199 - 200, 216, 230 designated under low confidence range (70 > pLDDT >50). Finally, amino acid range 144 - 168, 170, 203 - 215, 217 - 229 categorized under very low confident range (pLDDT <50).
Another major output parameter which analyses the predicted structure was PAE, which assess the inter domain accuracy. The region marked with dark green depicted as higher confidence with expected position error (Ao ) range from 0Ao to 31Ao .
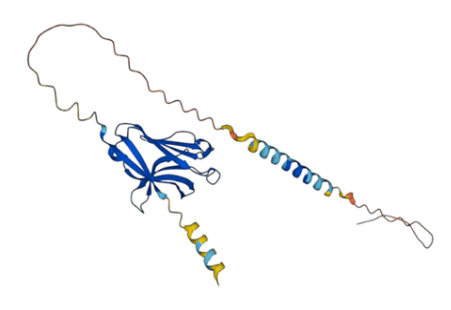
Figure 8: AlphaFold generated TREM2 3D Structure.
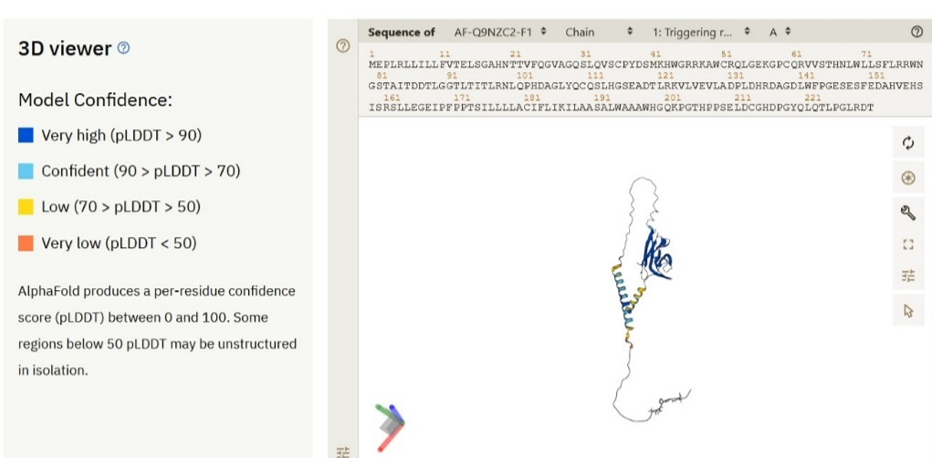
Figure 9: Local distance difference test (pLLDT) of TREM2 3D.
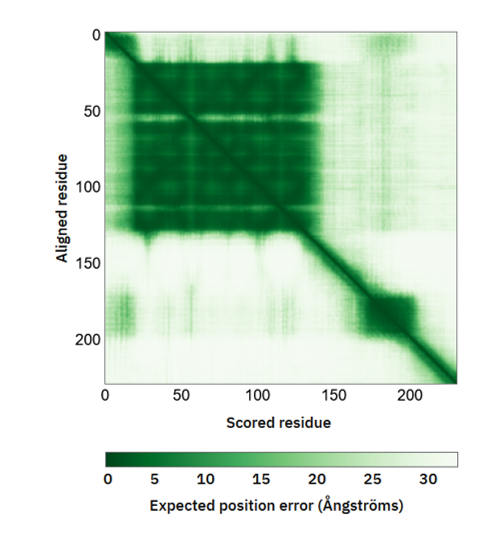
Figure 10: Predicted aligned error (PAE) of TREM2.
I - TASSER generates the decoys i.e. larger ensembles of structural confirmations of R47H TREM2. SPICKER program of I - TASSER clusters the decoys of R47H TREM2 variant. The top 5 models of R47H TREM2 were generated Figure 11 (a-e). The rankings of the same were depends upon the cluster size. C - score of the top model of R47H TREM2 was 0.15, estimated TM score was 0.73 ± 0.11 and estimated RMSD value was 3.9 ± 2.6 Ao. C - score of remaining top models were represented in (Figure 12). The best 10 threading templates of R47H TREM2 utilized by I - TASSER were depicted in (Figure 13). Other results were also generated by I - TASSER comprising of predicted secondary structure of R47H-TREM2 (Figure 14); predicted solvent accessibility of R47H-TREM2 (Figure 15); top 10 Identified structural analogs of R47H-TREM2 in PDB (Table 4); ligand binding site of R47H-TREM2 (Table 5) and enzyme commission number and active sites (Table 6).
ClusPro Molecular Docking analysis: TREM2-Aβ ligands interaction: ClusPro web server generates protein-protein interaction models of TREM2-Aβ ligands Figure 16 (a-f) and the results suggests that Aβ 6 has the highest binding affinity with TREM2 receptor. The top docking model of TREM2 - Aβ 6 complex exhibits cluster size with 337 members. The top model exhibits other docking outputs: Weighted score at center - 605.1: Lowest Energy - 654.7. The cluster size members of top models of remaining ligands in the descending order, along with its weighted scores were; Aβ 40 cluster size: 154; weighted score - center (- 1230.1); lowest energy (- 1230.1). Aβ 42 cluster size: 125; weighted score - center (- 1254.7); lowest energy (- 1390.4). Aβ 42 B cluster size: 121; weighted score - center (- 1366.1); lowest energy (- 1658.1). Aβ 42 A cluster size: 108; weighted score - center (- 1188.4); lowest energy (- 1188.4). Aβ oligomers cluster size: 101; weighted score - center (- 968.8); lowest energy (- 1214.0). The cluster size and weighted scores of all the TREM2 - Aβ ligands models were represented in the (Table 7 (a-f).
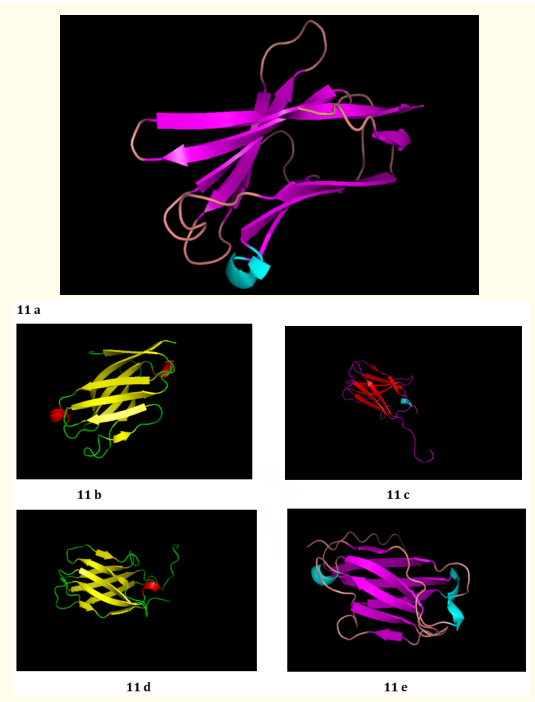
Figure 11: (a - e): Top Model (Model 1), 2, 3, 4 and 5 of R47H - TREM2 3D structure prediction by I-TASSER.

Figure 12: C - scores of R47H-TREM2 Models (1-5).
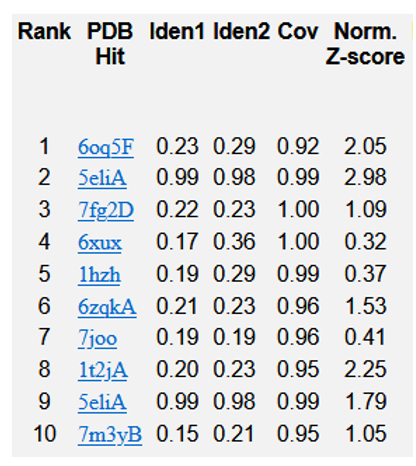
Figure 13: Top 10 templates of threading utilised by I-TASSER for R47H-TREM2.
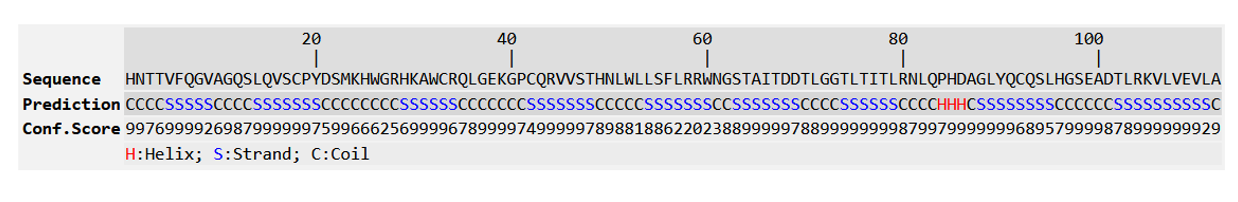
Figure 14: Predicted Secondary structure of R47H-TREM2.
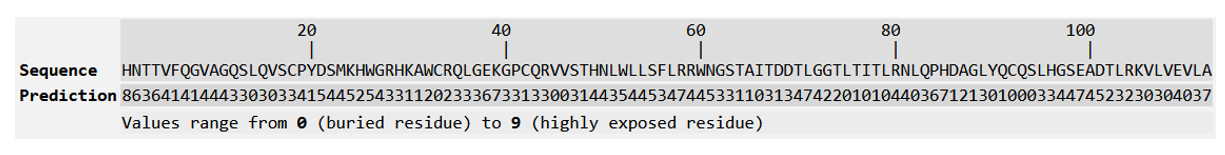
Figure 15: Predicted Solvent accessibility of R47H-TREM2.
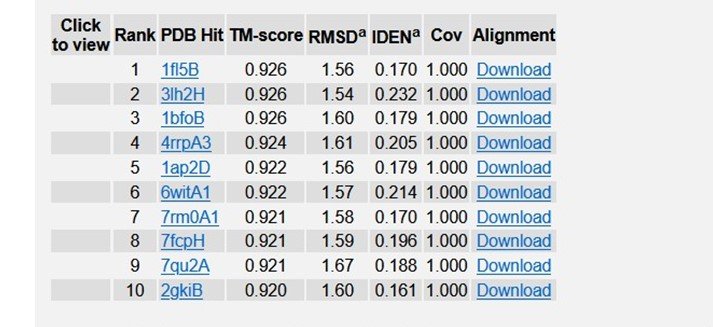
Table 4: Top 10 Identified structural analogs of R47H - TREM2 in PDB.
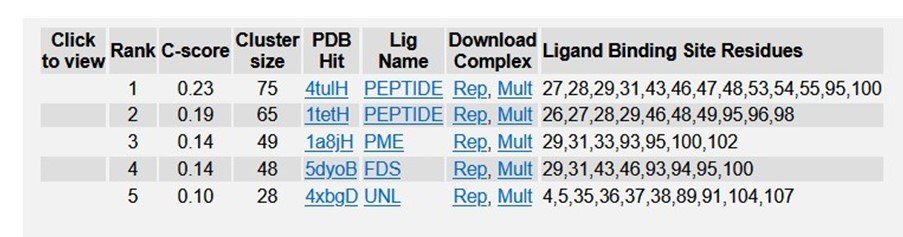
Table 5: Ligand binding sites of R47H - TREM2 in PDB.
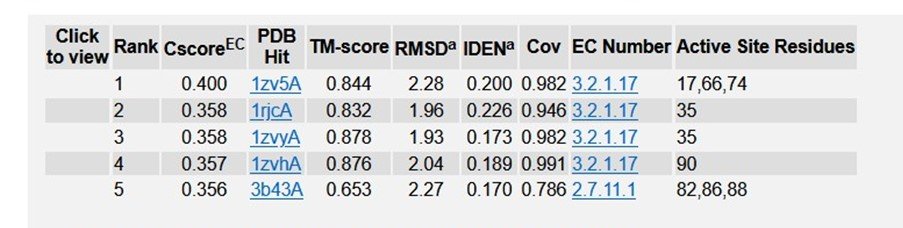
Table 6: Enzyme Commission (EC) numbers and active sites of R47H - TREM2 in PDB.
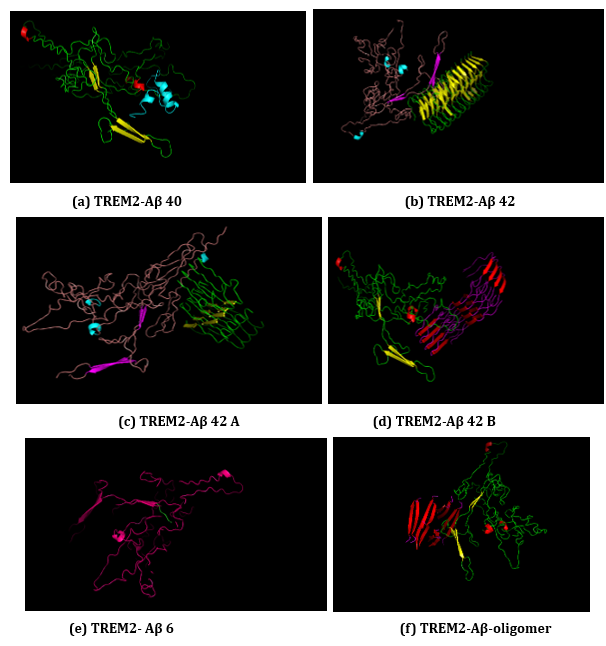
Figure 16: (a-f): ClusPro Molecular Docking analysis: TREM2-Aβ ligands interaction.
Do you watch television?
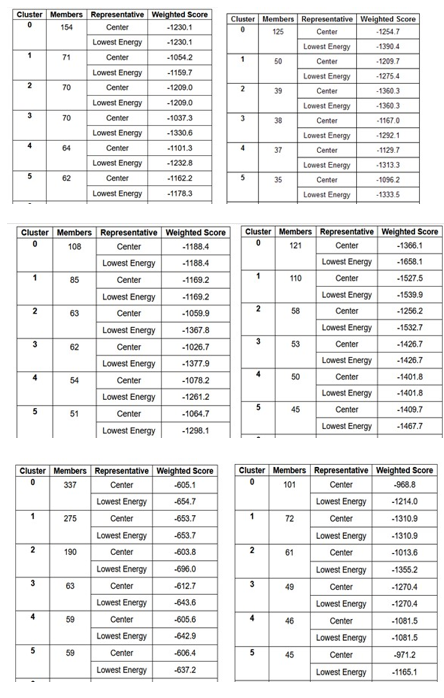
Table 7 (a-f): ClusPro based cluster size and weighted scores of TREM2-Aβ ligands interaction.
(a) TREM2-Aβ 40
(b) TREM2-Aβ 42
(c) TREM2 - Aβ 42 A
(d) TREM2 - Aβ 42 B
(e) TREM2 - Aβ 6
(f) TREM2 - Aβ oligomer.

Figure 17: HawkDock based TREM2-Aβ oligomer interaction: (a) TREM2-Aβ oligomer interaction - Model 1; (b) TREM2-Aβ oligomer interaction-GBSA analysis.

Figure 18: HawkDock based TREM2-Aβ 40 interaction: (a) TREM2-Aβ 40 interaction - Model 1; (b) TREM2-Aβ 40 interaction-GBSA analysis.

Figure 19: HawkDock based TREM2-Oligomer (3q9h) interaction: (a) TREM2 - Aβ Oligomer (3q9h) interaction - Model 1; (b) TREM2 - Aβ Oligomer (3q9h) interaction - GBSA analysis.

Figure 20: HawkDock based TREM2-Oligomer (3q9j) interaction: (a) TREM2 - Aβ Oligomer (3q9j) interaction - Model 1; (b) TREM2 - Aβ Oligomer (3q9j) interaction -GBSA analysis.
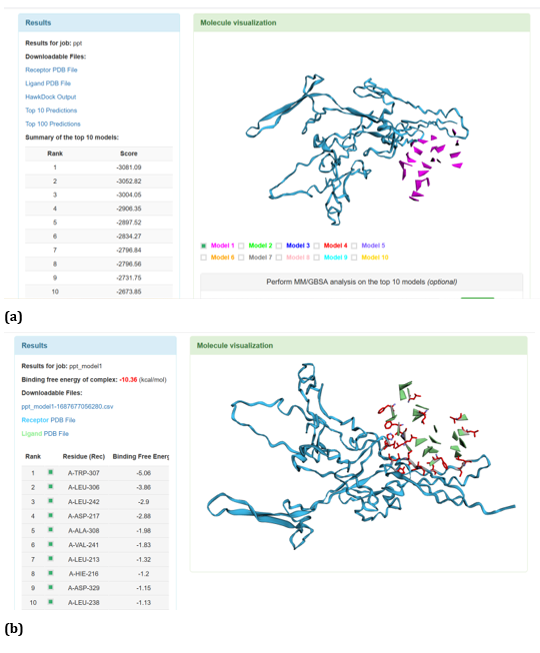
Figure 21: HawkDock based TREM2-Oligomer (3q9i) interaction: (a) TREM2 - Aβ Oligomer (3q9i) interaction - Model 1; (b) TREM2 - Aβ Oligomer (3q9i) interaction - GBSA analysis.

Figure 22: HawkDock based TREM2- Aβ 6 interaction: (a) TREM2 - Aβ 6 interaction - Model 1; (b) TREM2 - Aβ 6 interaction - GBSA analysis.
HawkDock protein-protein interaction analysis revealed that contradicting the ClusPro results, Aβ oligomer emerged as the favourable ligand for TREM2 with the highest score of - 4818.30 exhibited by top rank model of TREM2 - Aβ oligomer docking interaction. Additionally, the generalized born surface area (GBSA) analysis revealed that TREM2 - oligomer complex model exhibited binding energy of - 1.81 (kcal/mol) (Figure 17 (a, b). Docking scores of top models of other Aβ ligands in the descending order, along with its binding free energy were: Aβ 40: score: - 4524.58 and binding fee energy of the docked complex: - 66.07 (kcal/mol) (Figure 18 (a, b). Aβ oligomer (PDB ID-3q9h): score (-3502.48); binding free energy of the docked complex: - 13.72 (kcal/mol) (Figure 19 (a, b). Aβ oligomer (PDB ID-3q9j): score (-3155.89); binding fee energy of the docked complex: - 11.42 (kcal/mol) (Figure 20 (a, b). Aβ oligomer (PDB ID-3q9i): score (-3081.09); binding free energy of the docked complex: - 10.36 (kcal/mol) (Figure 21 (a, b). Aβ 6: score (-1587.19); binding free energy of the docked complex: - 17.89 (kcal/mol) (Figure 22 (a, b).
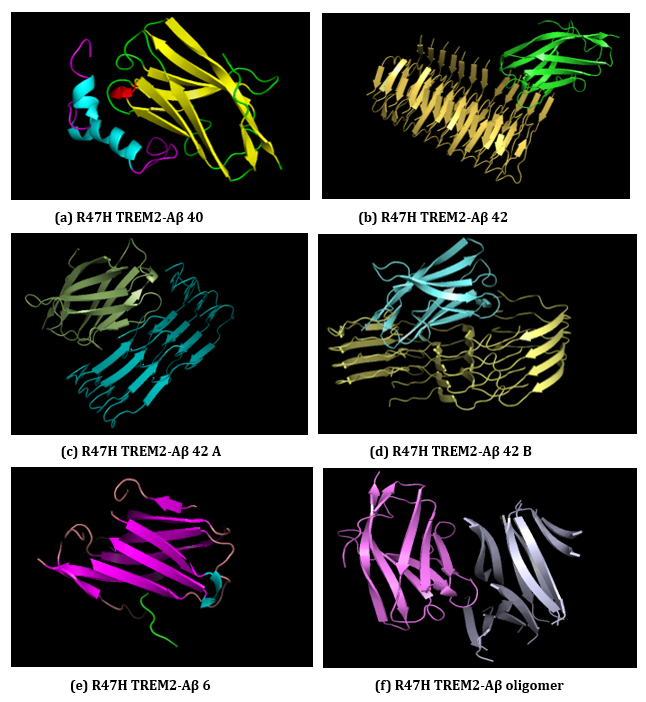
Figure 23: (a-f): ClusPro Molecular Docking analysis: R47H-TREM2-Aβ ligands interaction.
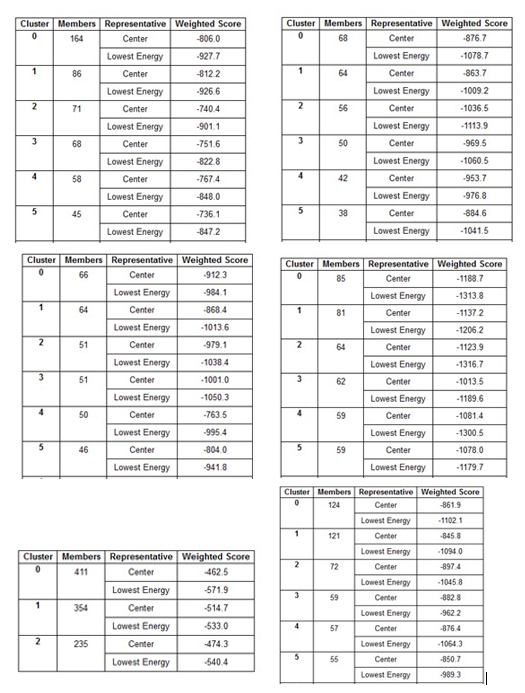
Table 8 (a-f): ClusPro based cluster size and weighted scores of R47H-TREM2-Aβ ligands interaction.
(a) R47H TREM2-Aβ 40, (b) R47H TREM2-Aβ 42, (c) R47H TREM2-Aβ 42 A, (d) R47H TREM2-Aβ 42 B
(e) R47H TREM2-Aβ 6, (f) R47H TREM2-Aβ oligomer.
ClusPro web server generates protein-protein interaction models of R47H-TREM2-Aβ ligands (Figure 23 (a-f) and the results generated through ClusPro suggests that Aβ 6 ligand has the highest ligand binding affinity with R47H TREM2 exhibiting a cluster size of 411 members. The weighted scores of R47H TREM2 - Aβ 6 interaction were at center - 462.5; lowest energy - 571.9. The cluster size members of top models of remaining ligands in the descending order, along with its weighted scores were Aβ 40; cluster size - 164; weighted score: center - (- 806.0); lowest energy - (- 927.7). Aβ oligomer; cluster size -124; weighted score: center - (- 861.9); lowest energy - (- 1102.1). Aβ 42 B; cluster size - 85; weighted score: center - (- 1188.7); lowest energy - (- 1313.8). Aβ 42; cluster size - 68; weighted score: center - (- 876.7); lowest energy - (- 1078.7). Aβ 42 A; cluster size - 66; weighted score: center - (- 912.3); lowest energy - (- 984.1). The cluster size and weighted scores of all top models of R47H - TREM2 - Aβ ligands were represented in (Table 8 (a-f).
With respect to R47H TREM2 - Aβ ligands docking, HawkDock protein-protein interactions studies suggests that contradicting the ClusPro results, Aβ oligomer emerged as the favourable ligand for R47H TREM2 with the highest score of - 4142.15 exhibited by top rank model of R47H TREM2 - Aβ oligomer docking interaction. Additionally, the generalized born surface area (GBSA) analysis revealed that the binding free energy of the R47H TREM2 - Aβ oligomer docked complex was - 32.33 (kcal/mol) (Figure 24 (a, b). Docking scores of top models of other Aβ ligands in the descending order, along with its binding free energy were: Aβ 40: docked score: -3142.34; binding free energy of the docked complex: -27.57 (kcal/mol) (Figure 25 (a, b). Aβ oligomer (3q9h): docked score: -2804.76; binding free energy of the docked complex: - 13.21 (kcal/mol) (Figure 26 (a, b). Aβ oligomer (3q9j): docked score: -2799.18; binding free energy of the docked complex: -1.87 (kcal/mol) (Figure 27 (a, b). Aβ oligomer (3q9i): docked score: -2640.56; binding free energy of the complex: 9.1 (kcal/mol) (Figure 28 (a, b). Aβ 6: docked score: -1308.43; binding free energy of the docked complex: -17.07 (kcal/mol) (Figure 29 (a, b).
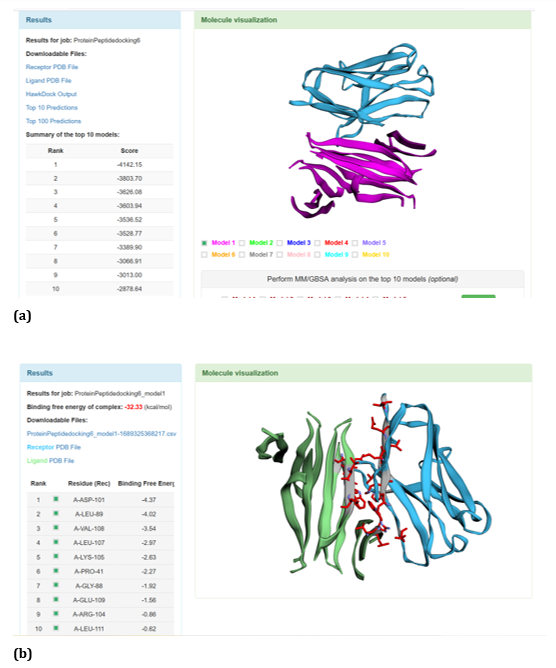
Figure 24: HawkDock based R47H-TREM2-Aβ oligomer interaction: (a) R47H-TREM2-Aβ oligomer interaction - Model 1; (b) R47HTREM2-Aβ oligomer interaction-GBSA analysis.

Figure 25: HawkDock based R47H-TREM2-Aβ 40 interaction: (a) R47H-TREM2-Aβ 40 interaction - Model 1; (b) R47H-TREM2-Aβ 40 interaction-GBSA analysis.

Figure 26: HawkDock based R47H-TREM2-Aβ Oligomer (3q9h) interaction: (a) R47H-TREM2-Aβ Oligomer (3q9h) interaction - Model 1; (b) R47H-TREM2- Aβ Oligomer (3q9h) -GBSA analysis.

Figure 27: HawkDock based R47H-TREM2-Aβ Oligomer (3q9j) interaction: (a) R47H-TREM2-Aβ Oligomer (3q9j) interaction - Model 1; (b) R47H-TREM2- Aβ Oligomer (3q9j) -GBSA analysis.

Figure 28: HawkDock based R47H-TREM2-Aβ Oligomer (3q9i) interaction: (a) R47H-TREM2-Aβ Oligomer (3q9i) interaction - Model 1; (b) R47H-TREM2- Aβ Oligomer (3q9i) -GBSA analysis.

Figure 29: HawkDock based R47H-TREM2-Aβ 6 interaction: (a) R47H-TREM2-Aβ 6 interaction - Model 1; (b) R47H-TREM2 - Aβ 6 - GBSA analysis.
6 Aβ ligands were tested against TREM2 by utilising ClusPro and HawkDock protein-protein docking programs. Aβ 40, Aβ 6 and Aβ oligomer were tested in both the docking algorithms based on the pathological significance. On the lines of comparative docking assessment, similar docking pattern was conducted against R47H TREM2. The top docked model in ClusPro server suggests Aβ 6 has highest affinity of binding against TREM2 with cluster size of 337. In case of R47H TREM2, similar to TREM2, Aβ 6 has highest affinity of binding, exhibiting cluster size of 411 members in its top model which is higher than TREM2. Regarding HawkDock docking program, Aβ oligomer exhibited highest docking score of - 4818.30 against TREM2 in its top model, whereas top model of R47H TREM2-Aβ oligomer exhibited - 4142.15. Most importantly, in ClusPro, Aβ 6 exhibited as the potential Aβ ligand against TREM2 and its mutant variant R47H TREM2, whereas in HawkDock program Aβ oligomer exhibited as the potential Aβ ligand. Interestingly, In ClusPro docking program R47H-TREM2 - Aβ 6 interaction model is stronger than TREM2 - Aβ 6 model (Figure 30). Our computational study suggests Aβ 6 and Aβ oligomer has promising therapeutic potential in Alzheimer’s disease (AD). Further experimental investigation is required to decipher the role of Aβ 6 and Aβ oligomer in Alzheimer’s disease (AD).
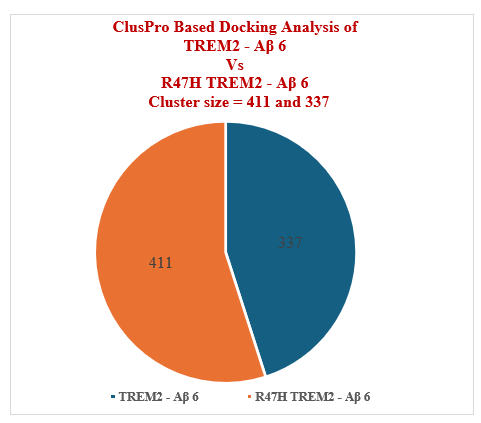
Figure 30: ClusPro Based Docking Analysis of TREM2 Vs R47H TREM2.
Alzheimer’s disease is a complex and insidious neurodegenerative disorder comprising heterogeneous pathophysiological characteristics [1]. Emerging research suggests that Triggering receptor expressed on myeloid cells 2 (TREM2) associated with higher risk of late onset AD (LOAD) [6]. TREM2 receptors alongside with its DAP12 adaptor proteins were indispensably required for phagocytosis, proliferation, survival and motility of microglia [9]. The signaling mechanism of the TREM2 receptor complex is still unclear [10]. Therefore, under this background, we have proposed in our previous paper [11] a hypothesis regarding TREM2 signalling pathway. Our Hypothesis states that TREM2 receptors of microglial cells along with its DAP adaptor proteins driven signalling cascade facilitates amelioration of Aβ aggregates in AD. In our hypothesis, we hypothesise that TREM2 ecto domain binds to both Aβ monomers and oligomers though with differential efficacy and we assume that Aβ oligomer has highest binding affinity with TREM2 ecto domain due to its high soluble nature. Two questions arises from our hypothesis, that, whether TREM2 has differential binding affinity with Aβ ligands? And To test whether Aβ oligomer has the highest binding affinity with TREM2 ecto domain? The current computational study was designed to ascertain our hypothesis. Comparative computational study of WT TREM2 and R47H TREM2 was performed to ascertain the pathophysiological role of R47H TREM2. To our knowledge, ours was the first of its kind study and we believe our computational investigation will give a novel insights for further experimental investigation and have huge therapeutic potential in AD. Computational based protein structure prediction and studying protein-protein interactions becoming crux point in construing various molecular and cellular mechanisms of imperative biological processes [19]. In the backdrop of expensive and tardy nature of experimental based structural determination of proteins [12] and as most of the important biological interactions occurs in transient complexes, computational tools explores novel horizons. Computational tools opens novel ways of target identifying mechanisms in the field of neurodegenerative disorders. ITASSER generates the top 5 3D models of TREM2. C - score of the top model of TREM2 was - 5, RMSD score: 20.4 ± 1.6 AO and TM score was 0.19 ± 0.04. With respect to R47H TREM2, output scores of top model of R47H TREM2 were C - score: 0.15, RMSD: 3.9 ± 2.6 AO and TM score was 0.73 ± 0.11. In order to analyse the structure of TREM2 based on different algorithmic framework, in addition to I-TASSER, we have used AlphaFold data base to generate 3D structure of TREM2. The major objective of employing different algorithms in structural analysis of TREM2 is to further explore the structural dynamics of TREM2. AlphaFold generates 3D structure of TREM2 under major output categories: predicted local distance difference test (pLDDT) and predicted aligned error (PAE). In pLDDT, different amino acids and amino acid ranges categorised under very high confident range (pLDDT > 90); high confident category (90 > pLDDT > 70); low confident range (70 > pLDDT > 50) and very low confident range (pLDDT < 50). PAE measures the inter domain accuracy. A family of 9 Aβ ligands were imported from PDB database: Aβ 40, Aβ 42, Aβ 42A, Aβ 42B, Aβ oligomer, 3 oligomer types of Aβ and Aβ 6. In ClusPro and HawkDock docking programs 6 ligands were tested. 3 Aβ ligands-Aβ 40, Aβ 6 and Aβ oligomer were commonly investigated in both ClusPro and HawkDock considering its pathological significance in AD. To ascertain the differential ameliorating ability of TREM2 and R47H TREM2 against various Aβ ligands, we have employed two different protein-protein docking algorithms-ClusPro and HawkDock. ClusPro program employed cluster size based ranking, whereas, HawkDock based on the binding energy. Varied TREM2 mutants results in detrimental pathological consequences. Kober and Brett [53] suggests that R47H, and R62H were the TREM2 variants which reduced the TREM2 binding ability. Mutations in TREM2 ecto domain (Y38C and T66M) leads to Nasu Hakola disease (NHD) and Fronto Temporal Dementia (FTD) [54]. As far as R47H TREM2 mutation is concerned, it is the chief cause of Late onset Alzheimer’s disease (LOAD) [55] and impairs the receptor-ligand interaction by disrupting the ligand binding domain [56]. Comparative structural studies of WT TREM2 and R47H TREM2 suggests that, in both the loops of complementarity determining regions (CDR) of TREM2, Arg-47 (R47) was the key amino acid determining the functional specificity of TREM2 binding to varied ligands [57]. Impaired binding of TREM2 - Aβ results to the Aβ plaque accumulation and weakening of immunity.
The protein-protein interaction results of ClusPro server suggests that Aβ 6 has the highest binding affinity with TREM2, with the top model exhibiting cluster size of 337 members. Similarly, with respect to R47H TREM2, ClusPro results suggests that Aβ 6 has the highest ligand binding affinity with R47H TREM2 with cluster size of 411 members. As per the ClusPro docking analysis, Aβ 6 is the best ligand for both TREM2 and R47H TREM2. Aβ 6 exhibited higher binding affinity against R47H TREM2 with 411 members when compared to TREM2 which exhibited 337 members.
Interestingly, HawkDock protein-protein interaction studies suggests that Aβ oligomer emerged as the favourable ligand for TREM2 with the top model exhibiting highest score of - 4818.30. HawkDock results of R47H TREM2 suggests that Aβ oligomer emerged as the best ligand for R47H TREM2 with highest score of - 4142.15. HawkDock investigation suggests that Aβ oligomer exhibited as the favourable ligand for both TREM2 and R47H TREM2, where in, Aβ oligomer exhibited better favourability against TREM2 in comparison with R47H TREM2. Our computational docking investigation suggests that Aβ 6 and Aβ oligomer as favourable ligands for both TREM2 and R47H TREM2 which requires further experimental investigation and have therapeutic potential in Alzheimer’s disease (AD). With respect to the first question of our hypothesis, the computational findings suggests that, in line to our hypothesis, TREM2 exhibited differential ligand binding ability, with highest binding affinity exhibited against Aβ 6 which is a monomer (ClusPro analysis) and Aβ oligomer (HawkDock analysis). Pertaining to the second question of our hypothesis that, whether Aβ oligomer has the highest binding affinity with TREM2? From the computational results, it can be deciphered that our results were in partial consensus with our hypothesis, as both Aβ 6 and Aβ oligomer exhibited as the potential ligands for TREM2. We believe that our computational investigation gave prudent insights for further exploration of TREM2- Aβ ligand interaction studies. Regarding R47H TREM2-Aβ ligand interaction studies, cluspro results suggests that Aβ 6 has better affinity with R47H TREM2 in comparison to TREM2, therefore, signifying potential therapeutic implications. HawkDock results suggests that affinity of Aβ oligomer with R47H TREM2 is close to that of TREM2, potentially signifying the therapeutic role of R47H TREM2.
The study which was closely related to our computational study was conducted by Mai., et al. [58]. Mai and colleagues conducted the docking molecular dynamic simulations between TREM2 and APOE protein. The ℇ4 isoform of APOE is considered as the most susceptible genetic risk variant for late-onset Alzheimer’s disease (LOAD). APOE exists in three isoforms ℇ2, ℇ3 and ℇ4. TREM2 -APOE interaction has modulatory effect on the AD pathogenesis. Their findings suggests that APOE isoform determines the strength of interaction with TREM2, where in, APOE ℇ4 has the highest binding energy with TREM2, followed by APOE ℇ3 and APOE ℇ2. From the results, it can be deciphered that despite APOE ℇ4 increases the susceptibility to AD, exhibited highest binding energy with TREM2. Further, R47H, which is a mutant variant of TREM2 diminishes the interaction ability of TREM2 with APOE, which may attribute to lower hydrogen bonding interaction, electrostatic forces and hydrophobic interactions between TREM2 and APOE.
Interestingly, studies of structural and biophysical aspects of R47H TREM2 suggests that R47H mutational variant does not impact the structure (or) stability of TREM2 protein, but it may disrupt the significant ligand interaction ability [56,59]. Based on this observation, we believe that R47H mutation doesn’t impact the structural stability of TREM2, therefore, may require further experimental study to decipher the mutant role in binding affinity.
The current computational work was conducted to test our hypothesis regarding differential Aβ ligand ability of TREM2 which has applications in Alzheimer’s therapy. R47H TREM2 docking studies was performed for corroboration. The overall objective of our computational investigation is construing of interaction of TREM2 with Aβ ligands and identifying potential Aβ ligand with promising therapeutic applications in Alzheimer’s disease. The study of differential docking analysis suggests that for both TREM2 and R47H TREM2, Aβ 6 and Aβ oligomer were exhibited as the potential ligands with promising therapeutic applications in Alzheimer’s disease (AD). R47H TREM2 increases the AD risk, our computational docking analysis suggests that it has highest binding affinity with Aβ 6 in comparison to TREM2 which also requires further experimental studies. Structural analysis of TREM2 based on different algorithms in the way of I-TASSER and AlphaFold explores further insights. Finally, our computational findings were in consensus with the first question of our hypothesis, in the way of TREM2 exhibiting differential ligand binding ability, with highest binding affinity exhibited against Aβ 6 and Aβ oligomer. Pertaining to the second question of our hypothesis, computational results were in partial consensus with our hypothesis, as both Aβ 6 and Aβ oligomer exhibited as the potential ligands for TREM2. We believe that our computational examination gave prudent insights for further exploration of TREM2- Aβ ligand interaction studies and have promising therapeutic potential against Alzheimer’s disease (AD).
SK Chand Basha is thankful and grateful to KLEF [KLU] Postdoctoral fellowship, to my parents, though they left me but their blessings are always with me, and also grateful to Almighty for guiding and showing the right path. SK Chand Basha is grateful to Prof. KS Jagannatha Rao, Pro chancellor of KLEF for his constant encouragement. M. Janaki Ramaiah is thankful to KLEF management for constant encouragement.
SCB designed, literature search, conceptualised, conducted structural modelling, docking, analysis and interpretation of results, and wrote the primary draft of the manuscript. MJR corrected the manuscript.
The current work was supported by KLEF [KLU] Post-doctoral fellowship awarded to SK Chand Basha.
SK Chand Basha is one of the members of Review Board of this journal, but was not involved in the process of peer review nor had access to any peer review information.
Data generated (Protein structure models and docked models) in support of the current work will be made accessible by the authors to any interested researcher.
Copyright: © 2024 SK Chand Basha and Mekala Janaki Ramaiah. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff







