TijanaVujanić Stankov1,2*, Aleksandar Ješić3, VojislavaBugarski Ignjatović1,2 Željka Nikolašević1 and Marija Semnic1,2
1Faculty of Medicine, University of Novi Sad, Novi Sad, Serbia 2Clinic of Neurology, Clinical centre of Vojvodina; Novi Sad, Serbia 3Medical Ward, Seychelles Hospital, Victoria, Seychelles
*Corresponding Author: TijanaVujanić Stankov, Faculty of Medicine, University of Novi Sad, Clinic of Neurology, Clinical Centre of Vojvodina, Serbia.
Received: February 22, 2021; Published: March 15, 2021;
Background: Cerebral hemiatrophy or Dyke-Davidoff-Masson Syndrome is caused by developmental hypoplasia or acquired atrophy of one cerebral hemisphere and leads to diverse clinical entities. In most cases, diagnose is made upon childbirth or during childhood period.
Case: We present a case of 30 years old woman who was admitted to our Clinic of Neurology with left-sided hemiparesis and a history of epileptic seizures during her childhood as well as perinatal hypoxia and reanimation after childbirth. Her magnetic resonance imaging exam showed right cerebral hemiatrophy. Further on, we discuss etiology, radiological morpho-functional findings and cognitive impairment in the patient with DDMS.
Conclusion: In mild cases, this diagnose of Dyke-Davidoff-Masson Syndrome could be made in adulthood. A diverse clinical presentation of this syndrome through lifetime could suggest that influence of both neuroplasticity in early childhood and brain-aging processes later in life on mildly, developmentally affected brain is unpredictable in many ways. Therefore, the significance of every presented case is important for clinicians as well as brain researchers.
Keywords: Right-sided Cerebral Hemiatrophy; Adult;Dyke-Davidoff-Masson Syndrome
DDMS: Dyke-Davidoff-Masson Syndrome; MRI: Magnetic Resonance Imaging; CT:Computed Tomography; AED:Antiepileptic Drugs.
Cerebral hemiatrophy, also known as Dyke-Davidoff-Masson Syndrome (DDMS) is a condition characterized by epileptic seizures, facial asymmetry, hemiplegia or hemiparesis and cognitive impairment [1]. It was first published as a series of nine patients by Dyke, Davidoff and Masson in the year of 1933. Regarding etiology, the disorder can be classified as congenital (primary) or acquired (secondary) [2,3]. When cerebral injury happens in utero, it is considered as congenital, like congenital malformation, infection and vascular abnormalities (coarctation in midaortic arch, ischemic and hemorrhagic states). Perinatal causes, like birth trauma, hypoxia or intracranial hemorrhage, and postnatal causes, like tumor, trauma, infection or prolonged febrile seizures, are considered as acquired DDMS [2,3]. The prognosis of this disease is better when the hemiparesis develops after the age of two in the absence of prolonged or recurrent seizures [2].
The diagnosis is based upon patient’s history, neurological exam, radiological, neuropsychological and EEG findings. Neurological exam could reveal facial asymmetry and ipsilateral hemiparesis or hemiplegia, language disturbances and/or low cognitive performance in one or multiple cognitive domains [1-3].
As for the radiological aspect of DDMS, computed tomography (CT) and magnetic resonance imaging (MRI) demonstrate the parenchymal abnormalities, unilateral loss of cerebral volume and/or compensatory bone alterations in the calvarium. All of the patients have unilateral cerebral hemiatrophy, which could be localized only in brain cortex or affect both subcortical and cortical brain structures. Additional neuroimaging findings could include prominent cortical sulci, thalamic or brainstem atrophy, atrophy of lentiform nucleus or corpus callosum, contralateral cerebellar atrophy, encephalomalacia, midline shift or/and porencephaly. The second characteristic of this syndrome are bone alteration like ipsilateral calvarial thickening, overdevelopment of paranasal sinuses, elevation of petrus ring, sphenoid wing and orbital roof and displacement of falx attachment [2-5]. Still, ipsilateral calvarial changes are not characteristic for acquired cases [2].
As for the neuropsychological profile, main characteristic of DDMS is global cognitive decline seen through higher cortical dysfunction dependent upon atrophy side. In patients with left-sided hemiatrophy, the emphasis has been given on language disorders with repetitive speech patterns and lack of spontaneity. In right-sided hemiatrophy, patients experience mainly visuospatial processing impairment, as well as attention, executive functions or memory impairment, without speech disturbances. Learning difficulties are also reported[1].
One of the hallmarks of this syndrome are epileptic seizures. The main seizure types are simple partial motor, complex partial motor and secondary generalized seizures. Interictal EEG includes prominent lower voltage and slow background activity in the atrophic hemisphere. Although usually separated by long intervals, seizures may change its character in time [1].
A 30 years old female patient referred to our Neurology Clinic with a history of left-sided hemiparesis with epileptic seizures during her childhood and without cognitive disturbances during her lifetime. At the childbirth, she suffered from hypoxia and she was resuscitated. From that time on, she had left brachioradial reflex lateralization and no other neurological consequences were noticed further on. At the age of ten, she experienced her first epileptic seizure, which started as left-sided hemiconvulsions with consecutive secondary generalization. The EEG made in that period showed slowing above right hemisphere. During next few years, she had three more left-partial motor epileptic seizures with secondary generalization, already under antiepileptic drugs (AED) with Phenobarbiton. Her further psychomotor development was without any difficulties. After her secondary education, where she experienced no learning difficulties, the patient started to work as a nurse. At the age of 24, when she got pregnant, she decided to stop taking AED. After her delivery, she started experiencing mild left sided weakness of her arm and leg, but did not consult the doctor immediately. Two years later, her first CT scan showed porencephaly in her right temporal lobe. At the age of 30 she was sent to examination to our Clinic.
At the neurological examination, mild spastic left hemiparesis was found. At that time, she was not receiving any AED. The MRI scan showed severe right cerebral hemiatrophy with dilated sulci frontally and right lateral ventricle, without any other brain region atrophy, nor any pathological bone changes (Figure 1-10).

Figure 1&2:T2 axial MRI scan showing cerebral hemiatrophy on the right side of the brain with temporal porencephalic cyst, with preserved subcortical structures.

Figure 3&4:T2 axial MRI scan showing cerebral hemiatrophy on the right side of the brain localized parietally, occipitally and frontally.and

Figure 5&6:T2 coronal MRI scan showing right sided hemiatrophy.

Figure 7&8:T2 coronal MRI scan showing right sided hemiatrophy (cerebellum coronal plate

Figure 9&10:T1 sagital MRI scan showing right sided hemiatrophy.
The MRI diffusion tenor imaging (DTI) showed no white matter brain alterations with normal arrangement of white matter tracts (Figures 11 to 14). The EEG featured non-specific electrocortical dysfunction (Figure 15).

Figure 11&12:MRI DTI tractography reconstruction at the level of capsula interna.

Figure 13&14: MRI DTI tractography reconstruction at the level of corona radiate and spelenium corporis callosi.
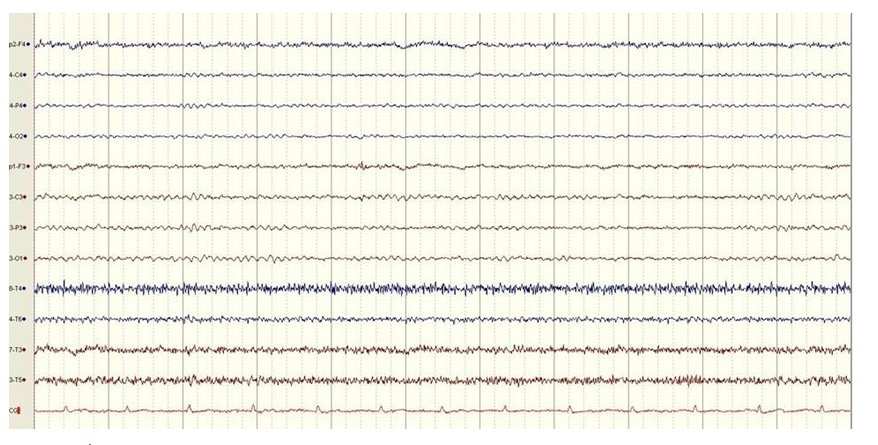
Figure 15:EEG in patient with DDMS.
Her neuropsychological assessment indicated the presence of a mild cognitive impairment (MCI) with signs of right hemisphere dysfunction, including subtle deficit in the visual-constructive and visuospatial organization in two dimensions (Figure 16) and deficit in visual memory, especially delayed recall of visual material (Figure 17). Additionally, there was evidence of attention deficit manifested by faster mental fatigue during prolonged cognitive demands.

Figure 16&17:Achievement on immediate and delayed visual reproduction recall from Wechsler Memory Scale Revised.
Cerebral hemiatrophy or DDMS is caused by developmental hypoplasia or acquired atrophy of one cerebral hemisphere, which is characterized by epileptic seizures, facial asymmetry, hemiplegia or hemiparesis and cognitive impairment [1,2]. Based upon etiology, this syndrome can be either congenital or acquired. Regarding the fact that this patient suffered from hypoxia at the childbirth and was resuscitated, the type of DDMS in this case would be acquired. As for her pyramid deficit developed at the age of 27, it is to be assumed that brain-aging processes could potentially be evidentearlier in life in patients with some brain structural abnormalities than in individuals without structural brain abnormalities.
Radiological characteristics of this syndrome consist of unilateral loss of cerebral volume and diverse spectrum of brain structure abnormalities [1]. The compensatory skull changes reflect adaptations to unilateral decrease of brain tissue, but calvarial compensatory changes are mainly not observed in acquired cases [2-4]. In this case, only right-sided hemiatrophy was dignosed, without any other brain region atrophy nor bone alterations. Shen., et al. described three MRI patterns of cerebral hemiatrophy: diffuse cortical and subcortical atrophy (I), diffuse cortical atrophy associated with expanded porencephalic cysts (II) and old infarction with necrosis in the territory of the middle cerebral artery (III). Patterns I and II are believed to have resulted from hypoxic-ischemic encephalopathy [5]. Since this patient had hypoxic injury at the childbirth and MRI revealed only cortical atrophy, her MRI finding is considered to be as pattern II. Furthermore, recently developed imaging techniques, like MRI DTI, could provide new insights into brain structure in patients with DDMS. In that manner, Sarikaya`s., et al. published the results on MRI DTI in patient with right-sided DDMS showing severe reduction in fractional anisotropy (FA) on the left corticospinal tract compared to the opposite, as well as decreased FA in contralateral superior cerebellar peduncle [4]. The MRI DTI finding in our patient showed no brain white matter alterations in both hemisphere, which is in accordance to her structural MRI, where no subcortical atrophy was described.
Even though the patient had no disabilities in activities of daily living, the neuropsychological assessment showed MCI with impairment in the field of visual-constructive and visual-spatial organization. This result is in accordance with previously reported small groups of DDMS patients where orientation discrimination tasks showed lower results in comparison to face discrimination tasks. The explanation probably lies in the fact that orientation relies on processing in the dorsal visual pathways localized in parieto-occipital lobe and it is associated with spatial vision which show right hemispheric lateralization, whereas facial discrimination leads to ventral bilateral activation [1]. Further neuroimaging studies could illuminate these underlying mechanisms. Additionally, disturbances are also noticed in visual memory tasks and attention. Since our patient had atrophy of right frontal, parietal and partially temporal lobes, the impairment of attention and visual memory, as signs of affected non-dominant brain hemisphere, is to be expected. In the end, the fact that our patient had no functional disabilities in her daily living activities in the presence of evident right-sided cerebral hemiatrophy support the neuropsychological view that psychological functions are diffusely represented in the right hemisphere and only significant structural damage to the right hemisphere is accompanied by greater neuropsychological deficits. Still, the neuroplasticity could also be the explanation in some extent of our patients’ neuropsychological achievement, since the hemiatrophy happened early in life. Therefore, more DDMS cases alike are necessary to be analyzed in the future.
The EEG abnormalities have been reported in individual cases with this syndrome. From our patients’ medical history we found that the EEG findings, when she was nine years old, showed slowing above her right hemisphere, which is characteristic EEG finding in this syndrome [1]. Since the patients had no epileptic seizures more than 15 years, she decided not to take AED and her control EEG showed non-specific electrocortical dysfunction. Despite the evidence of structural cortical brain alteration and history of prior epileptic seizures, the absence of recurrent seizures, even without any AED, are one of the major prognostic factors for the illness progression.
The differential diagnosis of DDMS includes conditions that are associated with cerebral hemiatrophy like Rasmussen encephalitis (RE), Sturge-Weber syndrome, Silver-Russell syndrome, basal ganglia germinoma and Fishman syndrome. Most of these can be differentiated by performing a thorough clinical examination and by neuroimaging [6]. The most challenging diagnose to be ruled out in this case was RE. As a chronic progressive immune-mediated disorder of children between 6 and 8 years of age, RE presents with intractable and recurrent focal epilepsy, cognitive impairment and similar imaging findings of hemispheric atrophy without any calvarial changes [7,8]. Since our patient had hypoxia and reanimation at childbirth, the probable etiology of DDMS in this case is vascular, rather than immune-mediated, like in RE cases. Additionally, progression of a disease in our patient with rarefication of epileptic seizures, without necessity of AED later in life, the slow progression of hemiparesis and the assumption, that cognitive impairment did not progress during more than 20 years of life, does not correspond to disease progression seen in RE patients. As for the other diagnosis of cerebral hemiatrophy, a proper clinical history could differentiate the diagnosis from DDMS.
The DDMS is not a very common disorder and not any kind of cerebral hemiatrophy should be named as DDMS. Still, based upon clinical features with pyramid deficit, different types of epileptic seizures in time, most of which with focal onset and secondary generalization and mild neuropsychiatric impairment of non-dominant hemisphere, this state is to be considered as right-sided Dyke-Davidoff-Masson syndrome and further neurological state of this patients is to be evaluated over time. Even though DDMS is mainly pediatricians diagnose, in mild cases, this diagnose could be made in adulthood. A diverse clinical presentation of this syndrome through lifetime could suggest that influence of both neuroplasticity in early childhood and brain-aging processes later in life on mildly, developmentally affected brain is unpredictable in many ways. Therefore, the significance of every presented case is important for clinicians as well as brain researchers.
None.
Citation: : Tijana Vujanić Stankov., et al. “Adult Right-sided Cerebral Hemiatrophy: Dyke-Davidoff-Masson Syndrome in a 30 Years-old Patient”. Acta Scientific Neurology 4.4 (2021): 07-12.
Copyright: © 2021 : Tijana Vujanić Stankov., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.









 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff