Siddak Dhaliwal, John C W Short and Kyoungtae Kim*
Department of Biology, Missouri State University, USA
*Corresponding Author:Kyoungtae Kim, Department of Biology, Missouri State University, USA.
Received: July 18, 2024; Published: August 12, 2024
Citation: Kyoungtae Kim., et al. “Domain Truncation and Point Mutations Unveil Potential Vps1 Recruitment Mechanisms". Acta Scientific Microbiology 7.9 (2024):14-25.
Dynamins play pivotal roles in various cellular processes, including membrane trafficking, endocytosis, and organelle dynamics. This study focuses on Vps1, a dynamin-like protein in Saccharomyces cerevisiae, involved in diverse intracellular trafficking pathways. We investigated subcellular localization of Vps1 and its functional domain's role in the recruitment of Vps1 to different organelles, including the endosome and late Golgi. Through molecular cloning, site-directed mutagenesis and microscopy-based approaches, we elucidate the contributions of specific Vps1 domains and amino acid residues to its subcellular localization. Our findings shed light on the intricate regulatory networks governing membrane trafficking and organelle dynamics mediated by Vps1, contributing to a comprehensive understanding of intracellular transport processes in yeast cells..
Keywords: Vps1; Dynamin-Like Protein; Vps10; Endosome; Cargo Retrieval
Dynamin, a 100-kDa GTPase, is involved in various cellular processes such as vesicle trafficking, vesicle scission, synaptic vesicle recycling, organelle division, viral resistance, and cytokinesis (Ekal., et al. 2023; Gundu., et al. 2022; Hu and Reggiori, 2023) [5,8,12]. It is an essential component of vesicle formation in clathrin-mediated endocytosis, synaptic vesicle recycling, caveolae internalization, and possibly vesicle trafficking in and out of the Golgi (Ford and Chappie, 2019) [6]. There are five domains identified in a classical dynamin, namely: an N-terminal GTPase domain, a middle domain, a GTPase effector domain (GED), a pleckstrin homology (PH) domain, and a C- terminal proline-rich domain (PRD). These domains are implicated in nucleotide hydrolysis, self-assembly into rings or spirals, stimulating GTPase activity, membrane binding, and containing Src-homology 3 (SH-3) bindi.ng sites, respectively (Hinshaw, 2000; Moustaq., et al. 2016) [11,19]. It is suspected that Dynamin-binding proteins attach to the PRD, enhancing its GTPase activity or targeting it toward the plasma membrane. Dynamin's self-assembled structure aligns with the notion that dynamin wraps around the endocytic budding vesicle's neck and performs membrane fission at the plasma membrane (Hinshaw, 2000; Takei., et al. 1998) [11,28].
Although it is not just the plasma membrane where dynamin performs this function, it is present in other locations within the cell and implicated in processes, including cytokinesis, export from the Golgi, caveolae-dependent endocytosis, icropinocytosis, and autophagy. In mammals, Dyn1, Dyn2, and Dyn3 are coded by three classical dynamin genes. Dyn1 or Dyn2 is expressed highly in neurons or expressed ubiquitously, respectively. Dyn3 is also expressed in the brain but at much lower levels than Dyn1 (Urrutia., et al. 1997) [30]. Dyn1 has undergone thorough studies and is recognized for its role as a mechanochemical enzyme, primarily responsible for inducing vesicle constriction and fission during endocytosis (Moustaq., et al. 2016) [19]. In yeast, classical dynamin proteins are absent. However, there are three dynamin-like proteins (DLPs), namely Dnm1, involved in mitochondrial fusion and fission, Mgm1 plays a role in maintaining the mitochondrial genome and inheritance, and Vps1 is involved in membrane trafficking and scission (Sesaki., et al. 2003) [24]. All these DLPs contain the N-terminal GTPase, the middle, and the GED domain. Vps1, like Dyn-2, is involved in several membrane fusion/fission processes. This data suggests that Vps1 operates like the classical dynamin in mammalian cells (Nannapaneni., et al. 2010) [20].
As mentioned above, Vps1 function is required in many membrane remodeling events in cells, including the transportation of various receptor proteins, such as carboxypeptidase Y (CPY)- Vps10 complexes, the latter is CPY receptor (Raymond., et al. 1992; Robinson., et al. 1988) [22,23] from the late Golgi to the late endosome via anterograde transport (Gurunathan., et al. 2002) [9]. These Vps10 the CPY receptor are then recycle back to the Golgi via retrograde traffic. It has been demonstrated that Vps10 vesicles are pinched from the late endosome by the action of Vps1, resembling the function of dynamin protein in mammals. Dysregulation of the reterograde transport by Vps1 deficiency leads to the abnormal accumulation of these receptor proteins in endosomal compartments (Lukehart., et al. 2013) [17].
Altogether, Vps1 emerges as a regulatory element that facilitates the movement of vesicles throughout various components within cells. Although it is known that Vps1 is involved in numerous membrane trafficking processes, highly precise interactions must exist between Vps1 and its binding partners to ensure effective trafficking. In recent efforts, it has unveiled novel binding partners of Vps1, associated with yeast membrane traffic, such as Ypt6, Vps51, Snc2, Vti1, and Chc1(Goud Gadila., et al. 2017) [7]. However, understanding these associations and their physiological implications requires further investigation.
Additionally, we are unaware of the exact mechanism involved in targeting Vps1 to the Golgi, late endosome, or the plasma membrane, and thus also requires further investigation. The current research seeks to identify the specific domain(s) and amino acid residue(s) within Vps1 responsible for its localization to both the endosome and the late Golgi. In our research, we directed our attention to Vps1, specifically focusing on assessing the potential role of distinctive domains in the recruitment of Vps1 to the late endosome and the late Golgi. We further extended our investigation to examine the impact of point mutations of the following four amino acid residues including R465D, P564A, S599D, and K591E, for these sites are suspected to potentially hinder the functional capacity of Vps1. Overall, our results propose that the full- length Vps1 is competent for its recruitment to the late endosome. In addition, two residues of Vps1, R464 and P564 are playing a key role in Vps1 association with the late endosome.
Saccharomyces cerevisiae BY4741 (Invitrogen) was used as wild type (WT) control (Table 1), with stock colonies maintained locally at -80°C. Working colonies were incubated from -80°C storage into YPD (Yeast Peptone Dextrose) complete culture media containing 1% yeast extract, 2% peptone, 2% D-glucose, and cultured with shaking at 30°C. The grown cells were harvested in the mid-log phase (OD600 0.6 – 0.9), unless called for otherwise (such as in One-step transformation, below). Auxotrophic selective media made from Yeast Nitrogen Base (YNB) (Sunrise Science Products, Cat #1500-250) and Complete Supplement Mixture (CSM) minus relevant amino acids for nutrient dropout (e.g. -Ura, -Leu, -Ura -Leu, etc.). The VPS1 knockout (vps1∆, KKY0352) background strain was maintained as WT and generated as described previously in (Banh., et al. 2017) [2] and (Woodman., et al. 2016) [31]. All recombinant Vps1 truncated domain strains described below were also maintained and cultured using these protocols.
Table 1: Strains used for this study.
Genomic template DNA was previously isolated from WT yeast using Epicentre® MasterPure™ Yeast DNA Purification Kit (Cat # MC85200) as previously cite (Banh., et al. 2017; Woodman., et al. 2016) [2,31]. Genes of interest (VPS1), domain truncations (Vps1 GTPase, Mid, GED, Mid + GED, PRD, and GTPase + Mid), and mutation variants (Vps1 R465D, P564A, K591E, S599D) were amplified using the Phusion® Green High-Fidelity DNA Polymerase Kit (Thermo Scientific, Cat # F534L) in accordance with manufacturer specifications. Purification of PCR products was performed using the Macherey Nagel NucleoSpin® Gel and PCR Clean-up kit (Ref. No. 740609.250). Host vector pRS416, with –Ura auxotrophy, under the control of a TEF overexpression promoter, was used for generating recombinant plasmids. Ligation of PCR inserts into host vector was done using T4 DNA Ligase (Thermo Scientific, Cat # EL0014). Recombinant plasmids were inserted into WT and vps1∆ strains via one-step transformation (Chen., et al. 1992) [3].
Table 2: Plasmid used in the study.
Bacterial transformation of the generated recombinant plasmids was performed with DH5α E. coli, or Stellar Competent Cells HST08 E. coli (Clonetech, Cat # 636763) where appropriate. Competent cells were obtained from -80˚C storage and thawed on ice for 20-30 min. Prepared auxotrophic selection agar plates, typically Luria broth containing ampicillin (LB + amp) in 100 mm x 15 mm Petri dishes (Thermo Scientific, Cat # FB0875712), were obtained from 4˚C and warmed in a 37˚C stationary incubator prior to plating cells, along with SOC medium (Mediatech, Inc., Cat # 46-003-CR). Generally, a minimum total mass of 100 ng of recombinant plasmid were added to 100-200 µL of competent cells, and the mixture was incubated on ice for 30 min. Each was heat shocked at 42˚C for 45 sec, and placed back on ice for 2 min. The cell mixture was filled to 500 µL with warmed SOC medium, and incubated with shaking for 1 hr. 30 min. For plating, 100 µL of cells were pipetted onto LB amp plates and spread with silica beads, then grown overnight in a stationary 37˚C incubator. Unless otherwise stated, a positive colony from each plate was selected and subjected to diagnostic restriction digest to verify the presence of plasmid.
Yeast one-step transformations (Chen., et al. 1992) [3] were performed with WT and vps1∆ strains to create strains (Table 1). Cells were streaked from -80˚C storage onto fresh agar, YPD or YPD + kanamycin, respectively, and grown for 24-48 hrs. Selected positive colonies were inoculated into liquid media and grown overnight with shaking at 30˚C to a target OD of 0.6 – 0.9. Cells were pelletized for 10 min at 1000 rpm, while transformation buffer (final concentration 0.2 M lithium acetate, 40% PEG, and 1X DTT) was prepared, filter purified, and kept on ice until used. Growth media was replaced with 100 µL transformation buffer, to which 5 µL of boiled and chilled salmon sperm DNA (Thermo Scientific, Cat # AM9680) and 400-500 ng of purified recombinant plasmid were added. The transformation mixture was vortexed vigorously, then heat shocked in a 45˚C water bath for 30 min. Following heat shock, cells were plated onto drop- out plates (SC-URA) with silica beads. Plates were incubated for 48 hrs., or until colonies appeared, in a stationary 30˚C incubator. Unless otherwise stated, one positive colony from each plate was selected and subjected to colony PCR to verify the presence of plasmid.
Membrane markers of three organelles were selected for genomic C-terminal GFP fusion. Endocytic sites were marked by actin-binding protein 1 (Abp1-GFP), Golgi bodies were marked by Golgi-localized, Gamma-adaptin ear homology, Arf-binding protein 1 (Gga1-GFP), and late endosomes were marked by vacuolar protein sorting protein 10 (Vps10-GFP). Tagging of Vps1 plasmid variants was achieved by fusing mRFP to its N-terminal side. C-terminal mRFP fusion with Vps1 has been shown to inhibit its polymerization and is therefore not useful for tagging Vps1 (Chi., et al. 2014) [4].
Primers were designed for the truncated expression of the three putative, modular domains of VPS1 in five variations, plus one “PRD-like” experimental domain, for a total of six variants, using SnapGene Viewer (GSL Biotech LLC): GTPase (KKP 656, 657), Mid (KKP 658, 659), GED (KKP 660, 661), GTPase+Mid (KKP 656, 659), Mid+GED (KKP 658, 661), and PRD-like (KKP 759, 760) (Table 3). Maintenance of codon integrity and the reading frame was ensured by referencing the ExPASy Translation tool (Swiss Institute of Bioinformatics, https://web.expasy.org/translate/). PCR products of VPS1 variants were cleaned up, digested, ligated, and fused with mRFP into TEF pRS416 (Table 2) as detailed under the MOLECULAR CLONING subheading. The recombinant vectors were transformed into DH5α E. coli, grown and isolated, and transformed into WT and vps1∆ yeast cells (KKY 0002, 0352) (Table 1).
(table 3)
Point mutations were introduced into VPS1 to change the character of selected residues of Vps1 (R465D, P564A, K591E, and S599D) (Table 3) using the Phusion Site-Directed Mutagenesis Kit (Thermo Scientific, Cat # F541, Lot # 00520116). Phosphorylated primers (Table 3) encompassing the swapped nucleosides were designed and purchased from IDT (Integrated DNA Technologies). Desiccated primers were received and diluted in 1 mL nuclease-free water (NF H2O) to establish stock solutions. Working solutions of primer reagent for use in PCR reactions were diluted from stock to 10 µM final concentration with NF H2O. Liner fragments were PCR amplified under normal conditions, creating linear PCR products with phosphorylated ends. The phosphorylated ends were ligated and circularized in a 5-minute ligation reaction with T4 DNA Ligase. The resulting circularized plasmids were transformed into DH5α™ Competent Cells (Thermo Scientific, Cat # 18258012), grown overnight in liquid LB, and miniprepped as described above. Samples of purified mutant plasmids were prepared to specifications and shipped to Eurofin Genomics for sequencing verification of induced point mutations. Following verification by sequencing, the purified mutant plasmids were digested with EcoRI and XhoI and ligated into the mRFP-fused position in TEF pRS416 as described above.

Table 3: Primers used in the study.
Colocalization assays were performed utilizing a spinning confocal fluorescence motorized inverted Olympus microscope (Model # Olympus IX81), with high-resolution DIC and phase contrast optics, equipped with an air-cooled Laser Launch System (Intelligent Imaging Innovations, Inc., Model # 3iL32) laser stack, with the GFP channel at 488 nm and the RFP at 561 nm. The microscope is coupled with an ImagEM charge-coupled device (EM-CCD) digital camera (Hamamatsu, Model # C9100-13), controlled by an ORCA camera controller (Hamamatsu, Model # C10600). The motorized stage system is a ProscanTM II (Prior Scientific, Model # H107), and encompassed within a temperature and humidity-controlled light occlusion box (Hitschfel Instruments, Inc.). The system is all under the control of Slidebook 6 software (Intelligent Imaging Innovations, Inc.).
Yeast cell strains were inoculated from 4˚C stock into liquid media and grown overnight with shaking at 30˚C to log-phase (0.6 – 0.9 OD) prior to imaging. Depending on the cell density in solution, approximately 2.0 – 2.5 µL was pipetted onto glass slides (Fisher Scientific, Cat # 12- 544-1), then a glass slide cover (Fisher Scientific, Cat # 12-544-10), was placed over the droplet gently from one side, with care taken towards reducing bubbles. If cells were mobile due to capillary action, too dense, too dilute, or found aggregating in more than one focal plane, then the slide was discarded and prepared again until optimal. The focus was set to equatorial in bright field mode. Physiological assays of Vps10-GFP utilized only single-channel GFP, while all others were taken with simultaneous dual channel GFP and RFP. Photographs of live cells were taken at 200 – 500 ms, with channel intensity of 0 – 200 (of 255) units, depending on the quality of signal. Only healthy, budded, isolated cells were quantified; cells that were dead, not budded, aggregated into clusters, or contacting other cells did not meet the criteria for quantification. Typically, a 1.5 – 2-fold excess of 30 cells were photographed, to ensure a minimum count of 30 cells per trial, in triplicate trials.
Vps1 does not colocalize with Abp1 the endocytic marker.
Abp1, or Actin-binding protein 1, is a highly conserved protein involved in endocytosis regulation in yeast (Kaksonen and Roux, 2018) [14]. On the other hand, Vps1, a dynamin-related protein in yeast, is implicated in membrane trafficking and scission events, particularly in endosomal traffic and retrieval (Hinshaw, 2000; Jimah and Hinshaw, 2019) [11,13]. One of the studies observed temporal co-localization of Vps1 with Abp1 at the endocytic site suggesting a potential interaction at the plasma membrane (Palmer., et al. 2015) [21]. Another research published demonstrated a co-localization of Vps1 with endocytic reporters Abp1 and Rvs167, supporting its localization to endocytic sites and late endosomes (Smaczynska-de Rooij., et al. 2015) [26]. However, another investigation reported rare overlap between Abp1-marked endocytic patches and Vps1, indicating a lack of significant co-localization (Kishimoto., et al. 2011) [15].
Similarly, recent findings from our lab support this observation, as we reported that mRFP-Vps1 does not coincide with Abp1-GFP and Sla1-GFP, another endocytic marker (Goud Gadila., et al. 2017) [6]. This finding challenges the notion of Vps1's exclusive presence at the plasma membrane and instead suggests its involvement in late endosomal processes (Goud Gadila., et al. 2017; Hayden., et al. 2013; Lukehart., et al. 2013) [7,10,17].
Further investigations are warranted to reconcile these contradictory findings and elucidate the precise subcellular localization dynamics of these proteins. We revisited these studies and conducted our own experiments to shed light on this controversy. We expressed Abp1-GFP and various truncated mRFP-Vps1 species: Vps1-GTP (aa 1-340), Vps1-Mid (aa 341-614), Vps1- GED (aa 615-704), Vps1-PRD (aa 530-633), Vps1 Mid+GED (aa 341-704), and Vps1 GTP+Mid (aa 1-614), along with mRFP-Vps1 full length (aa 1-704). Our experiments revealed that mRFP- Vps1 full length, Vps1-Mid, and Vps1-GED exhibited less than 1% co-localization with Abp1- GFP, while no co-localization was observed with other truncated species. Additionally, Vps1- GTP and Vps1-PRD appeared blurry and fuzzy under fluorescence microscopy, suggesting a potential mistargeting of those Vps1 mutants due to domain truncation. Further attempts to fuse domains, such as mRFP-Vps1 GTP+Mid and Vps1 Mid+GED, did not result in co-localization either. In accordance with these observations, we established that these results strongly indicate that Vps1 does not co-localize with endocytic markers like Abp1.
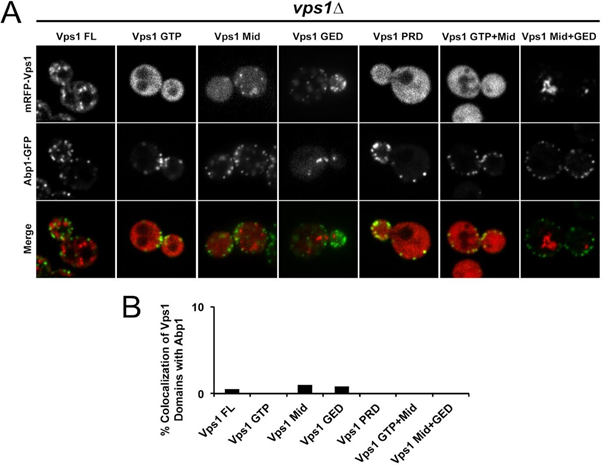
Figure 1: Level of co-localization between mRFP-Vps1 domains and Abp1-GFP. (A) mRFP- Vps1 full length and its truncated species in the first row. Abp1-GFP in the same strain in the second row. The third row shows the merged image. (B) Percentage of cells showing colocalization of mRFP-Vps1 domain to Abp1-GFP carrying endocytic dots.
Vps1’s partial association with Gga1
We then set out to study whether Vps1 domain(s) can be recruited to late Golgi marked by Gga1- GFP. Consistent with our prior observation (Goud Gadila., et al. 2017) [7], mRFP-Vps1 exhibited a modest level of co-localization with Gga1-GFP, accounting for less than 25% of the total observed cells (Fig. 2A). This finding suggests a partial association of full-length Vps1 with the trans-Golgi compartment. However, the degree of colocalization was relatively low, indicating that only a subset of Vps1 may be recruited to the Golgi.
In contrast, truncated Vps1 domains, namely Vps1-MID and Vps1-GED, showed significantly reduced colocalization with Gga1, with less than 5% of cells exhibiting overlap (Fig. 2B). This suggests a diminished ability of these truncated domains to be targeted to the trans-Golgi, possibly due to the loss of critical functional motifs or structural elements required for their targeting. Interestingly, no colocalization was observed between Vps1-GTP and Vps1-PRD domains with Gga1. Further, like previous experiments (Figure 1), Vps1-GTP and Vps1-PRD appeared blurry under fluorescence microscopy. Additionally, Vps1 truncated variants containing two domains, such as Vps1-GTP+MID and Vps1-MID+GED, did not result in colocalization with Gga1 either. Notably, Vps1-GTP+MID appeared fuzzy and blurry as well.
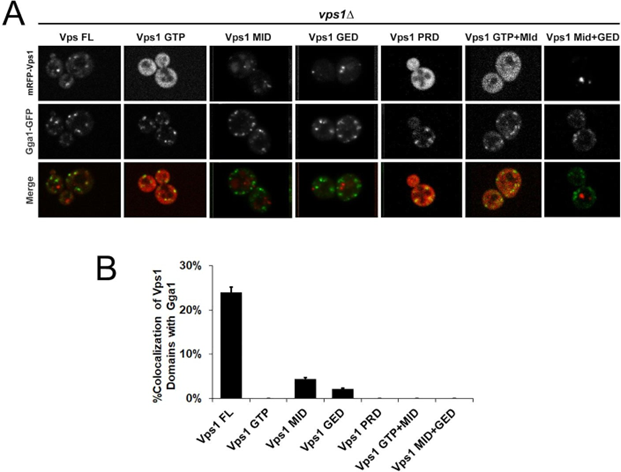
Figure 2: Level of co-localization between mRFP-Vps1 domains and Gga1-GFP. (A) mRFP- Vps1 full length and its truncated species in the first row. Gga1-GFP in the same strain in the second row. The third row shows the merged image. (B) Percentage of cells showing colocalization of mRFP-Vps1 domain to Gga1-GFP carrying endocytic dots.
We then investigated the extent of co-localization between mRFP-Vps1 species with Vps10-GFP at the late endosome. Notably, Vps1 full length showed co-localization with Vps10, approaching nearly 60% of observed cells as quantified in Fig. 3 A&B. However, none of the Vps1 truncated domains did exhibit colocalization with Vps10-GFP. Vps1 is widely known to retrieve Vps10- GFP from the late endosome/vacuole to late Golgi (Hayden., et al. 2013) [10]. Therefore, we anticipated observing Vps10-GFP accumulation at the late endosome/vacuole. Indeed, Vps1 KO mutant(∆vps1) and specific truncated Vps1 variants displayed a high percentage (approximately 80%) of Vps10-GFP accumulation at the endosome/vacuole, whereas Vps1 full length strain expressing mFRP-Vps1 exhibited less than 20% of cells with Vps10 accumulation at the vacuole compared to the wild type cells (0%), indicating a modest defect in Vps10 retrieval when wild type Vps1 is replaced with RFP-fused Vps1.
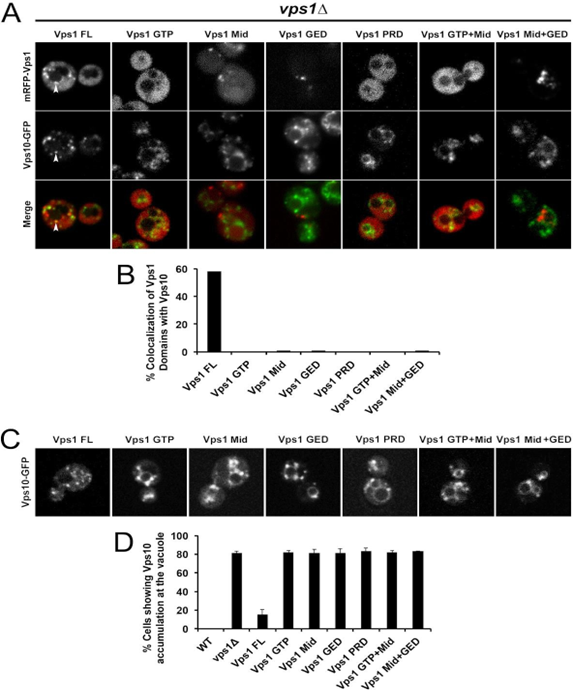
Figure 3: Level of co-localization between mRFP-Vps1 domains and Vps10-GFP. (A) mRFP- Vps1 full length and its truncated species in the first row. Vps10-GFP in the same strain in the second row. The third row shows the merged image. (B) Percentage of strains showing colocalization of mRFP- Vps1 domain to Vps10-GFP carrying endocytic dots. (C) Vps10-GFP distribution with respect to mRFP-Vps1 domain (D) Percentage of cells presenting Vps10-GFP accumulation at the rim of vacuole with respect to mRFP-VPS1 domain.
Next, we focused on assessing the extent of the colocalization between mRFP-Vps1 mutants carrying a point mutation each and Vps10-GFP in yeast cells; specific mutations were introduced to observe their effects on protein interactions. The mutated sites included Vps1 R465D, Vps1 P564A, Vps1 K591E, and Vps1 S599D in mRFP-Vps1 Full-Length.
In our investigation, the selected mutations (K591E, R465D, P564A, and S599D) aimed to elucidate the functional significance of the key residues in Vps1-mediated cellular processes. The K591 is a known PH binding site (Anand., et al. 2012) [1], thereby offering the opportunity for exploring the impact of binding site on Vps1 recruitment. Since, Vps1 binds PI3P (Hayden., et al. 2013) [10] and R465 is a presumable PI3P lipid binding motif (Misra., et al. 2001) [18], the main purpose of the R465D substitution was to elucidate the significance of PI3P binding in Vps1 function and membrane interactions. The substitution of P564A was constructed as the residue is presumed to be an SH3 protein binding motif (Kurochkina and Guha, 2013) [16] that is implicated in Vps1 function as suggested in a previous study (Smaczynska-de Rooij., et al. 2012) [25]. Finally, S599 residue was selected for investigation based on prior studies demonstrating its role as a key phosphorylation site regulating Vps1 function. A previous paper by Smaczynska-de Rooij., et al. (2015) [26] showed that phosphorylation at the S599 residue disrupts Vps1's ability to catalyze membrane scission during endocytic vesicle formation. Therefore, to better understand the role of phosphorylation dynamics in Vps1 function and recruitment, we replaced serine with aspartic acid.
Our findings revealed that only K591E displayed no significant divergence from the wild type, whereas the remaining three mutant variants, R465D, P564A, and S599D, exhibited a notable decrease in co-localization levels between Vps1 and Vps10-GFP. This suggests a crucial role for these sites in Vps1's recruitment. Particularly, the Vps1 P564A mutant exhibited significantly decreased co-localization levels with Vps10-GFP, with Pearson coefficient value <0.5, substantially lower than that of wild-type Vps1.
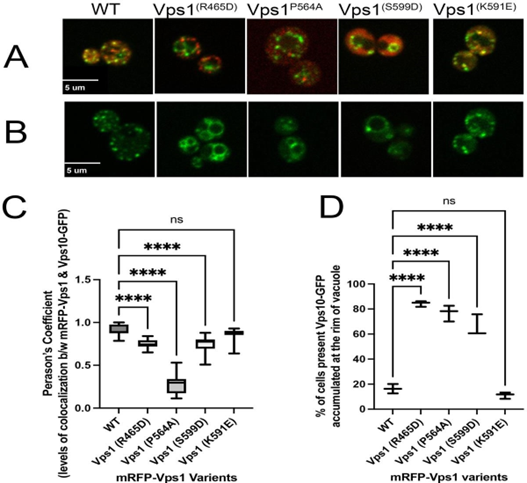
Figure 4: Level of co-localization between mRFP-Vps1 variants and Vps10-GFP and its impact on distribution of Vps10-GFP within the cell. (A) Cells showing mRFP-Vps1 mutated variants and Vps10-GFP during retrograde vesicular transport from late endosome to Golgi (B) Vps10- GFP distribution with respect to mRFP-Vps1 variants. (C) Levels of colocalization between mRFP-Vps1 variants and Vps-10 GFP measured in terms of Pearson’s coefficient and (D) Percentage of cells presenting Vps10-GFP accumulation at the rim of vacuole with respect to mRFP-VPS1 mutant variants.
The exact Vps1 recruitment to the endosome has been poorly understood. This study provides significant contributions to understanding the mechanisms behind Vps1 recruitment and its functional implications in endosomal cargo retrieval processes. Firstly, our investigation revealed that the K591 residue in Vps1 is not essential for Vps1’s recruitment, suggesting the involvement of alternative residues in this process. Secondly, we found that PI3P phospholipid at the endosome is relevant for Vps1 targeting based on our observation that the substitution of R465, a presumable PI3P binding residue, with aspartic acid disrupted Vps1 recruitment. This finding is consistent with the previous observations by Suzuki., et al. (2021) [27] in that in the absence of Vps34, a class III phosphatidylinositol 3-kinase (PI3K) responsible for generating PI3P on endosomal membranes, led to defects in the recruitment of Vps1. This dependency underscores the possibility of PI3P in mediating the recruitment of Vps1 to the endosome.
The phosphorylation site S599, when replaced with aspartic acid, exhibited reduced Vps1 co- localization at the endosome, suggesting the importance of dynamic phosphorylation and de-phosphorylation events for Vps1 recruitment. Interestingly, the lipid binding assays conducted by Smaczynska-de Rooij., et al. (2015) [26] showed that both wild-type Vps1 and the Vps1 S599D mutant exhibited the strongest binding to phosphatidylinositol monophosphate lipids, particularly PI3P. The S599D mutation caused a subtle but reproducible reduction in Vps1's interactions with PI4P and PI5P, compared to the wild-type protein. However, the S599D phosphomimetic mutation did not appear to affect Vps1's ability to bind PI3P (Smaczynska-de Rooij., et al. 2015) [26]. This suggests that the reduced co-localization of the S599D mutant with Vps10 cargo may not be solely due to impaired PI3P binding but could involve other mechanisms.
Mvp1, an SNX-BAR protein in yeast cells crucial for endosomal processes, recognizes the YXTXXFM motif on the endosomal protein Vps55 and forms oligomers through its PX and BAR domains. This oligomerization promotes cargo sorting into recycling vesicles, which subsequently invaginate into the cytoplasm. These vesicles are then pinched off the endosome by Vps1, facilitating cargo transportation to their final destination(Suzuki., et al. 2021) [27]. The authors proposed that dynamin-like GTPase Vps1 recruitment to the endosome is by interacting with the Mvp1 oligomers. Regarding Vps1 recruitment mechanisms, our results provide new insights that differ from the proposed model of Mvp1-dependent targeting. In their study when Vps1-GFP was expressed in vps35Δ snx4Δ mvp1Δ cells, it displayed endosomal localization; however, this punctate endosomal localization was lost in cells expressing the oligomerization mutant (Mvp1R346E/Q468E/W496E) with the same genetic background, indicating the requirement of Mvp1 oligomerization for Vps1 recruitment to the endosome (Suzuki., et al. 2021) [27]. Shockingly, Vps1-GFP maintained its endosomal localization in mvp1∆ cells. Therefore, although Mvp1 oligomerization is important for Vps1 recruitment, Vps1 recruitment to the endosome may not strictly depend on Mvp1. These observations that Vps1 can localize to the endosome in mvp1∆ cells suggest existence of alternative, Mvp1-independent mechanisms for Vps1 recruitment as well.
We propose a potential mechanism involving the P564 residue, which likely interacts with SH3 domain adaptor proteins, possibly facilitating direct or indirect interaction with phospholipids at the endosomal membrane. This suggests a novel mechanism for Vps1 recruitment, wherein the Vps1 adaptor must possess an SH3 domain for binding to the phospholipids at the endosome.
However, it is essential to acknowledge the limitations of our study, such as the focus on yeast cells as well as including the four untested ubiquitination sites on Vps1, warranting further exploration in future research. Together, our findings offer valuable insights into the intricate mechanisms governing Vps1 recruitment and shed light on its functional significance in endosomal processes.
We found that Vps1 is primarily recruited to the late endosome. Interesting, none of its truncated mutants was able to be targeted to the late endosome, regardless of the size of the truncated mutants, suggesting that only the full length of Vps1 is efficiently be targeted to the membrane. In addition, we concluded that the following three amino acid residues, R465, P564, and S599, play an important role in Vps1 targeting to the membrane to confer the corresponding Vps1 function in retrieving Vps10 from the late endosome.
Copyright: © 2024 Kyoungtae Kim., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff







