Kafayat Aderonke Yusuf and Kyoungtae Kim*
Department of Biology, Missouri State University, United States of America
*Corresponding Author: Kyoungtae Kim, Department of Biology, Missouri State University, United States of America.
Received: February 19, 2021; Published: March 25, 2021
Chemotherapy is one of the most effective treatment plans for several cancer types. The recurrent side effects derived from chemotherapy agents have warranted the search for novel chemical compounds with better efficacy and minimal side effects. In line with this idea, we investigated effects of a group of newly synthesized metal based chemical compounds called cyanoximates on HeLa human cancer cells. Cyanoximates used were Pt(DECO)2, Pt(MCO)2, and Pd(DECO)2 along with the chemotherapy drug Cisplatin as a positive control. We found that the metal cyanoximates reduced cell viability via apoptosis, and that Pt(DECO)2 was most effective among these new cyanomixates. In an attempt to understand the potential mechanism of action of Pt(DECO)2, we performed RNAseq analysis with HeLa cells treated with 0.5 mM Pt(DECO)2. Hundreds of genes in Pt(DECO)2-treated cells were differentially expressed with several upregulated genes known to be involved in cell cycle regulation and apoptosis. Our analysis also revealed that cancer growth promoting genes alongside drug transporter genes are downregulated in the presence of Pt(DECO)2. Taken together, our results provide evidence that Pt(DECO)2 can be an alternative agent for Cisplatin-based chemotherapy, and further, our transcriptomic analysis offers new insights into the mechanism of action of Pt(DECO)2 against cancer cells.
Keywords: Cancer; Chemotherapy; Cisplatin; Cyanoximates; Apoptosis; p53; HeLa Cells
Cisplatin: Cis-diamminedichloroplatinum; Pt(DECO)2: Bis (2-cyano-2-oxiamino-N,N’-diethylaminoacetamide) Platinum; Pt(MCO)2: Bis (2-oxiamino-2-cyano-N-morpholyl-acetamide) Platinum; Pt(DECO)2: Bis (2-cyano-2-oxiamino-N,N’-diethylaminoacetamide) Palladium; DMEM: Dulbecco's Modified Eagle Medium; DMSO: Dimethyl Sulfoxide; DHE: Dihydroethidium; DHR123: Dihydrorhodamine; ROS: Reactive Oxygen Species; PBS: Phosphate Buffer Saline.
Cancer can be illustrated as a group of diseases caused by abnormal growth and division of cells [1]. Cancer has been proclaimed as one of the world’s most complicated diseases with no permanent cure in sight for several tumor types, and millions of new cases reported every year [1]. The use of various metal-based compounds in chemotherapy is dated to ancient times due to their therapeutic properties [2]. The dawn of platinum-based compound “Cisplatin” in 1960 by Barnett Rosenberg [3] was a major improvement in the use of metal based compounds in the treatment of cancer cells and was considered the foundation for development of modern day metal-based anticancer drugs [3]. Carboplatin and Oxalilplatin are analogues of Cisplatin that have also been used for treatment of selected cancer types [4]. One major difficulty in the use of these chemicals is the multiple side effects as well as resistance of cancer cells to the treatment over time [5]. It is, therefore, pertinent to discover a new range of active metal-based compounds that haves minimal side-effects and can overshadow the long-term resistance of cancerous cells [6].
Cyanoximates are metal complexes of cyanoximes; the common name for a subclass of oximes that has been described as outstanding ligands with unique biological properties such as growth regulation, antimicrobial and detoxification activities [6,7]. It is fascinating to note that combination of the anticancer effect of platinum or palladium ions together with the proven therapeutic effects of cyanoxime ligands marked the starting point of cyanoximates preparation [8]. It is also noteworthy to state that the anti-proliferative activity and cytotoxicity of cyanoximes as well as their metal complexes have not been extensively studied, with only few existing publications about the use of cyanoximates in the treatment of cancer [6,8,9].
The present study is aimed at investigating the potential of newly synthesized cyanoximates, specifically Pt(DECO)2 and Pd(DECO)2, in the treatment of cervical cancer (HeLa cancer cells), using Cisplatin as control. Several studies suggest that most chemotherapeutic agents exhibit cytotoxic effects by reducing cell viability, triggering oxidative stress and instigating cell death [10]. In this regard, we assessed the effect of Pt(DECO)2 and Pd(DECO)2 on cell proliferation, cellular reactive oxygen species, and apoptotic cell death. We also employed the use of RNAseq analysis to quantify the transcriptomic alteration in cyanoximate treated cells. Finally, we postulated a model that describes the mechanisms of cyanoximate-induced toxicity in HeLa cells.
Cryopreserved human HeLa S3 cells (ATCC, VA) were thawed in a 37°C water bath. The cell line was cultured in Gibco Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (Gibco) and 1% penicillin and streptomycin antibiotics solution (Corning, NY). The cultures were then maintained at 37°C in a humidified 5% CO2/95% air atmosphere.
TreatmentAll chemicals tested in the present study were obtained from Dr. Nikolay Gerasimchuk from the Chemistry Department at Missouri State University. Cis-diamminedichloroplatinum (Cisplatin, Sigma-Aldrich), and Bis (2-oxiamino-2-cyano-N-morpholyl-acetamide) platinum (Pt(MCO)2) [8] were used for control experiments. Additionally, cells with no form of treatment were established as non-treated control. Bis (2-cyano-2-oxiamino-N,N’-diethylaminoacetamide) platinum (Pt(DECO)2) and Bis (2-cyano-2-oxiamino-N,N’-diethylaminoacetamide) palladium (Pd(DECO)2) [11] were used as major treatment for the experiments. All these chemicals were dissolved in dimethyl sulfoxide (DMSO, Fischer Scientific.) immediately before use. A DMSO treated control was also prepared.
Cell viability assayThe extent of viability of HeLa S3 cells treated with these chemicals was evaluated using XTT assay KIT (Biotium, CA). A total of 10,000 cells/well were seeded on a 96-well plate (Corning, NY) and incubated at 37°C for 24 hr prior to the treatment with varying doses of these chemicals, ranging from 0.125-1 mM, in tetraplicate for 24 hr. Then, XTT solution containing the XTT activator PMS in a 200:1 ratio was applied to each well, after which the plate was read on an ELx808 Absorbance Microplate Reader (BioTek, VT) for absorbance at A450-A630nm. The absorbance values at 5 hr after XTT treatment were collected.
Reactive oxygen species (ROS) measurementDihydroethidium (DHE) and Dihydrorhodamine 123 (DHR123, DHR) (Biotium, CA) that measure for superoxide and peroxynitrite levels, respectively, were used for this experiment. This investigation was carried out according to the previously described procedure [12]. To prepare stock solutions, DHE and DHR were initially dissolved in DMSO to get the desired concentration (10 mM stock). On the day of the experiment each stock was diluted to yield a 10 μM working concentration using 1X phosphate buffered saline (PBS). A total of 50,000 cells/well was seeded on a 24-well plate (Corning, NY) and cultured for 24 hr. The cells were then treated with 0.1 mM of chemicals indicated in the section above for 24 hr. On day 3, the cells were initially centrifuged to form a pellet, followed by treatment with freshly prepared ROS indicator dyes of 10 μM concentration. Each tube was treated with 1 mL of the dye solution and incubated for 30 minutes in the dark before analyzing with a flow cytometer (Attune NxT acoustic focusing cytometer, Life Technologies). The excitation and emission wavelength set for detecting oxidized DHE and DHR was 518nm/606nm and 507nm/536nm, respectively.
Apoptosis measurementApoptosis measurement was conducted as described by the manufacturer’s protocol (Thermo Fischer Sci., https://bit.ly/2mSZfhR). On day 1 of the experiment, 50,000 cells/well were seeded on a 24-well plate. Subsequently, the cells were treated with 0.1 mM of chemicals for 24 hr. On day 3, the cells were treated with 100 μL of 1X Annexin V binding buffer, 5 μL of Annexin V-APC dye (BD Pharmingen), and 5 μL of Propidium Iodide (BD Pharmingen). The cells were then incubated in the dark for 15 minutes and were subsequently analyzed using Attune NxT acoustic flow cytometer (Life Technologies). Annexin V-APC was excited by a laser line at 650nm, and the resulting emission wavelength of 660nm was recorded. The excitation and emission wavelength of propidium iodide was 533nm and 617nm, respectively.
Total RNA extractionOn day 1 of the experiment, HeLa cells were grown for 24 hr in a 6-well plate (Corning, NY) at a density of 1.0x106 cells per well. The cells were then treated either with 1% DMSO (non-treated control group) or 0.5 mM Pt (DECO)2 for 24 hr. RNA extraction was performed on day 3 using a TRIzol protocol (Invitrogen). The total RNA concentration was calculated using the Qubit 3.0 Fluorometer (Thermo Fischer Scientific). Final concentrations of total RNA ranged from 150-250 ng/μL. Two μg of total RNA for each sample was sent to the genome sequencing facility (University of Kansas Medical Centre, KS) for mRNA isolation and cDNA synthesis. mRNA isolation and cDNA synthesis
At the genome sequencing center, Stranded mRNA-Seq was performed using the Illumina NovaSeq 6000 Sequencing System. Quality control on RNA submissions was completed using the Agilent TapeStation 4200 using the RNA ScreenTape Assay kit (Agilent Technologies 5067-5576). Total RNA (1µg) was used to initiate the library preparation protocol. The total RNA fraction was processed by oligo dT bead capture of mRNA, fragmentation, reverse transcription into cDNA, end repair of cDNA, ligation with the appropriate Unique Dual Index adaptors, strand selection and library amplification by PCR using the Universal Plus mRNA-seq library preparation protocol (NuGEN 0508-08, 0508-32).
Library validation was performed using the D1000 ScreenTape Assay kit (Agilent Technologies 5067-5582) on the Agilent TapeStation 4200. Using the Agilent D1000 assayed library concentration, each library was diluted to ~4 nM and a final library quantification was conducted, in triplicate, using the Roche Lightcycler96 with Fast Start Essential DNA Green Master (Roche 06402712001) and KAPA Library Quant (Illumina) DNA Standards 1-6 (KAPA Biosystems KK4903). Using the qPCR results, RNA-Seq libraries were adjusted to 2.125 nM concentration and pooled for multiplexed sequencing. Pooled libraries were denatured with 0.2 N NaOH (0.04 N final concentration), neutralized with 400 mM Tris-HCl, pH 8.0. A dilution of the pooled libraries to 425 pM was performed in the sample tube on the instrument, followed by onboard clonal clustering of the patterned flow cell using the NovaSeq 6000 S1 Reagent Kit (200 cycle) (Illumina 20012864). A 2x101 cycle sequencing profile with dual index reads was completed using the following sequence profile: Read 1 - 101 cycles x Index Read 1 - 8 cycles x Index Read 2 - 8 cycles x Read 2 - 101 cycles. Following collection, sequence data was converted from bcl file format to fastq file format using bcl2fastq software and de-multiplexed into individual sequences for data distribution using a secure FTP site or Illumina Base Space for sequence analysis (University of Kansas Medical Center Genomics Core).
Analysis of sequencing dataData from the above-mentioned cDNA sequences was analyzed using Basepair tech, a website created for analyzing sequenced data (www.basepairtech.com). The data obtained from Kansas Medical Genome Center was uploaded to the server using the pipeline RNA-seq. Expression count (STAR) was generated for each data set, which comprised of a quality check to ensure that each file of the sequenced data had good quality reads and the data were interpreted correctly. Next, the reads were aligned to the human reference genome used (human hg19 genome) in order to get a list of differentially expressed genes (DEGs). After obtaining the gene data, genes with a q-value greater than 0.05 were not included in the final list of analyzed genes. The selected genes were grouped based on correlating Gene Ontology (GO) terms obtained from GOrilla.
Statistical analysisStatistical analysis was carried out with GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA). Analysis and comparison of data from treatment and control groups was evaluated using one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test. Differences between groups were considered to be significant at a p value of <0.05. Data was represented as Mean ± S.D. A single * represents p < 0.05, ** represents p < 0.001, and *** represents p < 0.0002, and **** represents p < 0.0001.
Little research has been conducted to assess the potential cytotoxic effects of metal cyanoximates on cancer cells, except for a couple of reports illustrating the effects of Pt(MCO)2 and Pd(MCO)2 on HeLa cell viability. Those studies employed the use of Trypan Blue assay for cell viability quantification, revealing that the treatment of Pt(MCO)2 and Pd(MCO)2 on HeLa and WiDr cells results in decreased cell viability up to 16% and 28%, respectively [6,8]. The current study, for the first time, evaluated the effects of Pt(DECO)2 and Pd(DECO)2 on HeLa cell viability by measuring the absorbance of XTT tetrazolium at 450-630nm (Figure 1). HeLa cells were treated with the metal cyanomixates, ranging from 0.125 mM to 1 mM by a two-fold serial dilution. Concurrently, we varied the concentration of DMSO, the vehicle control from 10% to 1.25% in order to accurately assess the actual effect of the tested metal cyanoximates. With 1 mM of Pt(DECO)2, Pd(DECO)2, and Pt(MCO)2, our results showed significant reduction in cell viability extent to which only less than 25% of cells were viable after the treatment (Figure 1A). However, this marked decrease in viability can be largely attributed to the 10% DMSO used to solubilize each of these metal cyanomixates, based on our observation that 10% DMSO controls also reduced cell viability drastically (Figure 1A). It was found that with 0.5 mM of cisplatin or Pt (DECO)2 diluted in a final concentration of 5% DMSO, significantly reduced cell viability when compared with the 5% DMSO vehicle control (Figure 1B). The mean absorbance of cells treated with 5% DMSO alone was recorded at 0.3985 ± 0.012, significantly different from that of cisplatin (0.119 ± 0.0262, p < 0.0001) and that of Pt(DECO)2 (0.2713 ± 0.060, p = 0.004). At 0.25 mM of metal cyanoximates with 2.5% DMSO, Cisplatin caused reduced cell viability with an absorbance value of 0.295 ± 0.05, when compared with 2.5% DMSO alone (0.512 ± 0.177). At the same concentration, Pt(DECO)2 showed modest reduction in cell viability (0.384 ± 0.083), without statistical significance (p = 0.325) (Figure 1C). At the lowest concentration of 0.125 mM with 1.25% DMSO, Cisplatin affected cell viability with absorbance value of 0.605 ± 0.124, exhibiting significant difference (p = 0.0025) when compared with 1.25% DMSO used as control 0.825 ± 0.059 (Figure 1D). At the same concentration, none of metal cyanoximate-treated cells showed changes in cell viability. These results led us to the conclusion that Pt(DECO)2 and Pd(DECO)2 are not as effective as Cisplatin in reducing cell viability. Notwithstanding, Pt(DECO)2, among those new metal cyanoximates, was most effective to reduce cell viability even with its concentration at 0.25 mM.
Pt(DECO)2 and Pd(DECO)2 did not significantly affect ROSStudies have shown a positive correlation between the efficacy of chemotherapeutic anticancer drugs and elevation of reactive oxygen species (ROS) [10,13,14]. Several ROSs exist, but specifically, nitric oxide, superoxide and peroxynitrite have all been implicated in several cancer pathogeneses [15] and have also been targeted for cancer treatment [15,16]. However, there are no studies delineating the effect of metal cyanoximates on ROS alteration. This prompted us to test the potential impact of metal cyanoximates on alteration of ROS levels using two well-known ROS indicators, Dihydrorhodamine (DHR) and Dihydroethidium (DHE) that detects peroxynitrite and superoxide levels, respectively [17]. Our results showed that 0.1 mM of Cisplatin caused a significant reduction in peroxynitrite levels with the mean percentage of 38.57 ± 2.821 (p<0.0001) when compared with 1% DMSO control samples with the mean of 75.91 ± 2.821 (Figure 2, A AND B). In contrast, cell samples with metal cyanoximates, including Pt(DECO)2, Pd(DECO)2, and Pt(MCO)2 at their 0.1 mM concentration, showed no effects in altering peroxynitrite levels, comparable to DMSO control samples without statistical difference (Figure 2 A AND B). Based on our processed data from flow cytometry (Figure 3 A AND B), Cisplatin at its 0.1 mM concentration significantly elevated superoxide levels, with an average of 32.82% ± 1.293 (p < 0.0001). The mean of DMSO treated control stayed at 16.70 ± 5.650, whereas all tested metal cyanoximates caused no alteration in superoxide levels similar to that of DMSO treated control (Figure 3 A AND B). These findings suggest that 0.1 mM concentration of Pt(DECO)2 and Pd(DECO)2 do not show significant effects on production of ROS.

Figure 1: Measurement of cell viability using an XTT cell viability kit. A total of 10,000 HeLa cells per well were seeded on a 96 well plate. The cells were treated with varying amounts of metal cyanoximates 24hrs after plating. The total treatment time with those metal cyanoximates was 24 hrs prior to the measurement of the tetrazolium absorbance for each sample at A450nm-A630nm. A) The absorbance of treated cells at 1.0 mM concentration. B) The absorbance of treated cells at 0.5 mM concentration. C) The absorbance measurements at 0.25 mM. D) The absorbance measurements at the lowest treatment of 0.125 mM concentration. * represents p < 0.05, ** represents p < 0.001, and *** represents p < 0.0002, and **** represents p < 0.0001.
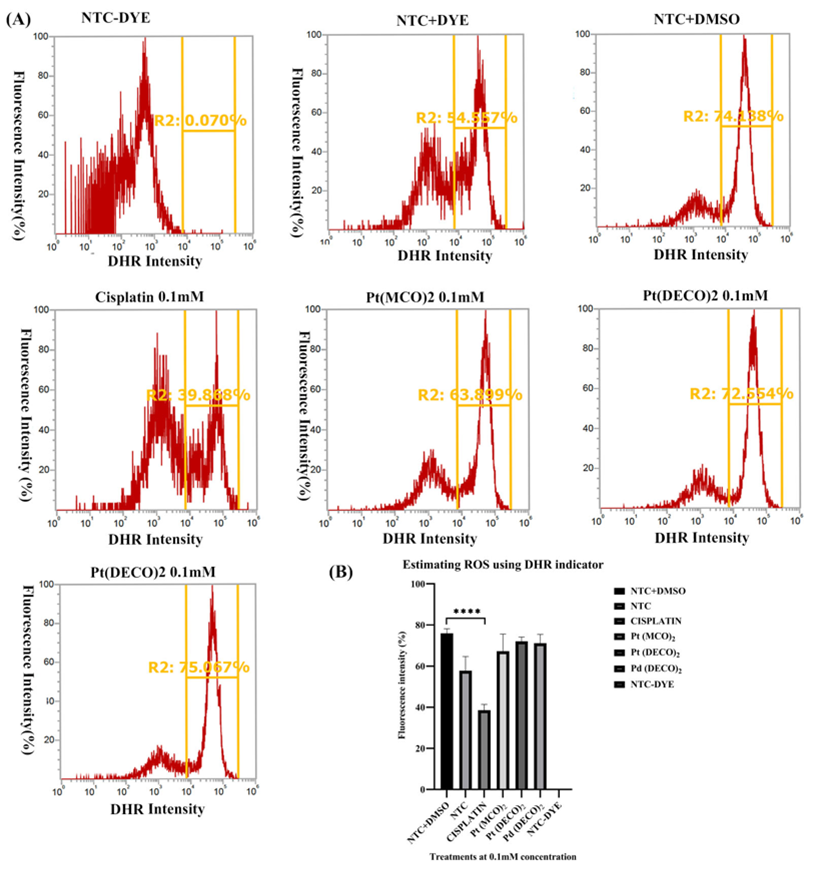
Figure 2: Measurement of peroxynitrite levels using DHR. On day 1, 50,000 cells per each well were seeded and cultured on a 24 well plate for 24 hrs. The cells were then treated with 0.1 mM of metal cyanoximates for 24 hrs prior to collecting them for the ROS analysis stated in the Materials and Methods section. Harvested cells were analyzed using the Attune Nxt Flow cytometer at excitation and emission wavelength of 507nm and 536nm, respectively. A) A representative flow cytometer image of each treatment and the corresponding gating used. Three replicates were used, but only one of the images is displayed. B) Analyzed data after using Prism. * represents p < 0.05, ** represents p < 0.001, and *** represents p < 0.0002, and **** represents p < 0.0001.
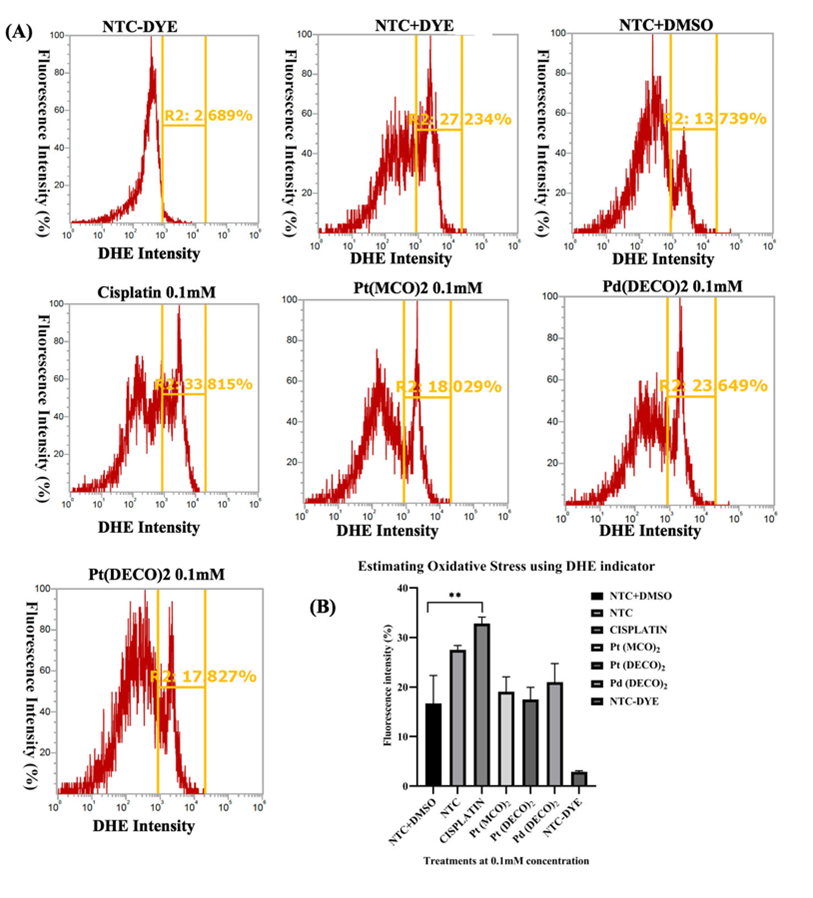
Figure 3: Measurement of superoxide levels using DHE. On the first day of the experiment, we seeded 50,000 cells per well onto a 24-well plate. The cells were then treated with metal cyanoximates for 24 hrs. On the third day, the cells were harvested for ROS analysis using the procedure described in the Materials and Method section. The cells were analyzed using Attune Nxt Flow cytometer at excitation and emission wavelength of 518nm and 606nm, respectively. A) A representative flow cytometer image of each treatment and the corresponding histogram gating used. Each treatment has exactly the same gating. Three replicates were used, but only one of the images is displayed. B) Analyzed data from Prism. * represents p < 0.05, ** represents p < 0.001, and *** represents p < 0.0002, and **** represents p < 0.0001.
Our metal cyanoximates including Pt(DECO)2 led to reduced cell viability, but not as effective as Cisplatin (Figure 1). Interestingly, however, the treatment with Pt(DECO)2 caused no alteration of ROS levels (Figures 2 and 3). In this regard, we hypothesized that the cell viability decrease with these metal cyanoximates may be attributed to significant levels of apoptosis induced by them. To validate our hypothesis, we exploited Annexin V-APC and Propidium Iodide that detect early and late stage of apoptosis, respectively [18,19]. With 0.1 mM of Pt(DECO)2, Pd(DECO)2, or Pt(MCO)2, our results showed increased levels of early apoptosis as Cisplatin (Figure 4, A AND B), significantly different when compared with DMSO control. However, levels of late apoptosis in treated cells were not distinctly different from control to DMSO control, because only less than 1% of cells in treated samples were in a late stage of apoptosis (Figure 4C). Elevation of early apoptosis with 0.1 mM of treated cyanoximates reveals that Pt(DECO)2 and Pd(DECO)2 can cause programmed cell death.
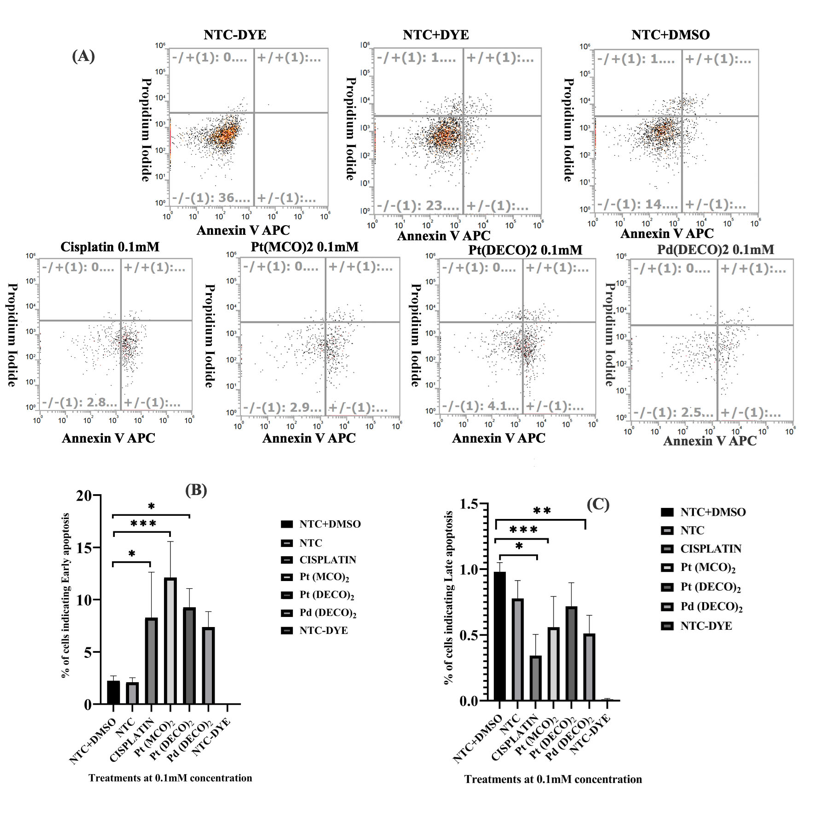
Figure 4: Measurement of HeLa cell death using Annexin V-APC and Propidium Iodide for early and late apoptosis, respectively. On day 1, 50,000 HeLa cells were seeded per well on a 24 well plate. The cells were then treated with 0.1 mM of metal cyanoximates for 24 hrs prior to collecting them for apoptosis measurements. On day 3, the cells were stained with Propidium Iodide and Annexin V-APC and were measured using a flow cytometer as detailed in the Materials and Method section. A) A representative flow cytometer image of each treatment and the quadrat gating used. The same gating was used for each treatment. The top left part of the quadrat shows necrotic cells, while the bottom left shows live cells. The top right quadrat shows late apoptotic cells, while the bottom right indicates early apoptosis. Three replicates were used, but only one of the images is displayed. B) Analyzed data after using Prism. Each bar represents the average of three replicates used with treatments showing early apoptosis. C) Analyzed data from Prism with treatments showing late apoptosis.* represents p < 0.05, ** represents p < 0.001, and *** represents p < 0.0002, and **** represents p < 0.0001.
Instead of concluding our studies based on data obtained from limited simple methods of assessing cell viability, reactive oxygen species, and apoptosis, we decided to conduct an RNAseq analysis in order to examine the global impact of the cyanoximates on HeLa cells. Due to our results in the previous section that demonstrate the potential of Pt(DECO)2 in inducing apoptosis, we examined the transcriptomic response in HeLa cells exposed to 0.5 mM concentration of Pt(DECO)2 by performing next-generation RNA sequencing that generates quantitative gene expression data for both the control and Pt(DECO)2 treated cells. The rationale for the increased concentration was that 0.1 mM was not sufficient to cause cell viability defects and that 1 mM of it was not possible due to the chemical’s solubility with low amount of DMSO, such as 1%. Briefly, the control and Pt(DECO)2 treated cells were subjected to total RNA extraction. Total RNA samples were then shipped to the genome sequencing center for mRNA isolation and conversion to cDNA. Both the control and Pt(DECO)2 treated samples were tested in triplicate, and the newly synthesized cDNA libraries were sequenced with a next-gen DNA sequencer (Illumina) that generates data set sequence for individual replicate. For differential expression analysis, each cDNA data set was uploaded and processed through the computational analysis platform (www.basepairtech.com), using the pipe line RNA-seq. The first process of analysis is to generate the expression count (STAR) on each data set. After trimming and processing, an average of 35,047,609 accepted reads were gathered from the DMSO (1%) control groups and 35,694,261 from the Pt(DECO)2 treated groups. Of these pure reads, an average of 97% and 95% of the total reads mapped to the human reference genome used (human hg19 genome) in the control and Pt(DECO)2 treated samples, respectively.
We identified that the expression levels of 1,703 genes in the Pt(DECO)2 treated samples were found to be statistically different (q<0.05) when compared to the non-treated control. Of all these significant genes, 869 genes were identified to be upregulated and 834 downregulated. From the gene pool, we obtained GO terms with GOrilla and found that 72% (627 of 869 genes) of the upregulated genes are responsible for cellular processes including, diffusion, energy production, cell transport, respiration, cell interaction, cell communication, and cell death. Likewise, genes implicated in metabolic processes constitute 51% of the upregulated genes (Figure 5A). A good number of the upregulated genes are also associated with regulation of gene expression (27%), transport (24%), catabolic processes (17%), translation (10%), protein localization (14%), and RNA processing (9%). In addition, 99, 49, 48, 16, and 3 upregulated genes found by the RNAseq analysis are known to be involved in cell response to stress, cytokine-mediated signaling pathway, protein ubiquitination, electron transport chain, and regulation of intrinsic apoptotic pathway, respectively (Figure 5A).
The GO term analysis on the downregulated genes reveals that regulation of biological activities is greatly downregulated at (69%) (Figure 5B). Other processes that were downregulated includes developmental processes (33%), regulation of cell communication (23%), transcription regulation (18%), and cell cycle processes (7%) (Figure 5B).
To further understand the cellular changes caused by Pt(DECO)2 treatment, we selected 190 most upregulated and 190 most downregulated genes, these genes have a log2fold change of at least 1.5. The vast majority of the highly upregulated genes are involved in multicellular organismal process (23%), and other highly upregulated process includes cornification or keratinization, regulation of gene expression, and gene silencing (Table 1). In the multicellular organismal process category, SOST and SOX3 genes are 5.4-fold and 5.6-fold upregulated, respectively. Several histone genes, including HIST1H3A known to be involved in multicellular organismal process, regulation of gene expression, gene silencing, nucleosome assembly, chromatin silencing, and chromatin silencing, are upregulated. HIST1H4B is also implicated in regulation of gene expression, gene silencing, nucleosome assembly, and chromatin silencing (Table 1). Our GO term analysis with 190 most downregulated genes showed that 52 out of 190 genes are involved in developmental processes. Other downregulated processes include signaling, regulation of synapse assembly, regulation of multicellular organismal process, and anatomical structure development (Table 2). NODAL (nodal growth differentiation factor gene) and SIX 1 (six homeobox 1 gene) appear to be involved in most of the downregulated processes, and both genes are involved in promoting tumorigenesis and transcription regulation (Kalyan., et al. 2017; Xia., et al. 2014) (Table 2).

Figure 5: Gene Ontology terms of differentially expressed genes (DEGs) with Pt(DECO)2. All DEGs were grouped into clusters of GO terms shown in the figure. A total of 1,703 DEGs were found to be statistically significant with q value below 0.05. A) Among 1,703 DEGs, 869 genes were found to be upregulated. B) The quantification of downregulated genes associated with their specific GO terms. Of the 1,703 DEGs, 834 genes were found to be downregulated.
| Gene Ontology Term | No of Genes | Corresponding genes |
|---|---|---|
Multicellular organismal process |
46 |
NPAS4, HIST1H3A, MOV10L1, CHRNA2, BRS3, FOXS1, KCNK3, KRT77, PCDH8, KRT3, SOST, CYP4F11, TRPM8, KRT37, SOX8, CACNA1S, JPH4, STC1, ACTC1, F2RL2, ANGPTL2, ALOX15, KRT34, ARHGDIG, ALK, MYH8, CRYAB, MYH6, KRT5, KRT6A, FGF4, KRT7, PLA2G2A, PPEF2, NRROS, SLC34A2, DAZL, SEZ6L, TRIM54, MYL9, RBM11, KRT72, HIST1H3F, NPTX2,KRT76, IRX4 |
cornification |
9 |
KRT5, KRT3, KRT6A, KRT72, KRT7, KRT34, KRT76, KRT37, KRT77 |
Keratinization |
9 |
KRT5, KRT3, KRT6A,KRT72, KRT7, KRT34, KRT37, KRT77, KRT76 |
Regulation of gene expression |
9 |
HIST1H4B, FAM172BP, HIST1H3F, HIST1H3A, MOV10L1, HIST1H4A, MIR31, MIR9-1, HIST1H1A |
Gene Silencing |
8 |
HIST1H4B, FAM172BP, HIST1H3F, HIST1H3A, MOV10L1, HIST1H4A, MIR31, MIR9-1 |
Hormone metabolic process |
7 |
ACE, SULT1E1, AKR1B10, ADH1C, RBP1, SPP1, AKR1B15 |
Nucleosome assembly |
7 |
DAXX, HIST1H4B, NAP1L2, HIST1H3F, HIST1H3A, HIST1H4A, HIST1H1A |
Cellular hormone metabolic process |
6 |
SULT1E1, AKR1B10, ADH1C, RBP1, SPP1, AKR1B15 |
Chromatin silencing |
5 |
HIST1H4B, FAM172BP, HIST1H3F HIST1H3A, HIST1H4A |
Actin filament-based movement |
4 |
MYBPC1, ACTC1, MYH8, MYH6 |
Actin-mediated cell contraction |
4 |
MYBPC1, ACTC1, MYH8, MYH6 |
Actin-myosin filament sliding |
4 |
MYBPC1, ACTC1, MYH8, MYH6 |
Chromatin silencing at rDNA |
4 |
HIST1H4B, HIST1H3F, HIST1H3A, HIST1H4A |
DNA replication-dependent nucleosome assembly |
4 |
HIST1H4B, HIST1H3F, HIST1H3A, HIST1H4A |
DNA replication-dependent nucleosome organization |
4 |
HIST1H4B, HIST1H3F, HIST1H3A, HIST1H4A |
Muscle filament sliding |
4 |
MYBPC1, ACTC1, MYH8, MYH6 |
Hormone catabolic process |
3 |
ACE, SULT1E1, SPP1 |
Table 1: Go term analysis with 190 top upregulated genes. A total of 190 upregulated genes with log2fold change of at least 1.5 were selected and analyzed using Gorilla (only 90 genes generated GO terms used in this table).
| Gene Ontology Term | No of Genes | Corresponding genes |
|---|---|---|
Developmental Process |
52 |
VASH2, NODAL, EXPH5, SRPK3, ADAMTS5, NRP2, SIX4, SEMA6A, MAP1A, HLF, ZDHHC15, GPM6B, LOX, P2RY1, SHISA2, TSSK4, EDAR, C6orf25, SLC1A3, ACTA2, TXNIP, JPH1, EYS, APLN, TNFRSF1B, ANKRD2, ADAMTS3, PLA2G3, SLFN5,KIF26B, AVIL, GDF15, ESCO2, E2F8, INHBE, PRICKLE1, FZD7, CRYGS, SIX1, RD3, GRIN1, ROBO2, RSAD2, GATM, SPRY1, GNAT2, SYNE4, ABCA12,CCDC39,NPY1R,PTPRD,DRAXIN |
Anatomical Structure Development |
40 |
ANKRD2,VASH2,ADAMTS3,NODAL,EXPH5,AVIL,SRPK3,GDF15,ADAMTS5,INHBE,E2F8,PRICKLE1,NRP2,SIX4,SEMA6A,MAP1A,HLF,CRYGS, SIX1, GPM6B, RD3, LOX, GRIN1,ROBO2,SHISA2, GATM,TSSK4, SPRY1, GNAT2, EDAR, C6orf25,SLC1A3, ACTA2, ABCA12, CCDC39, JPH1, EYS, APLN,TNFRSF1B, DRAXIN |
Regulation of multicellular organismal process |
40 |
ANKRD2,KCNIP4,VASH2,NODAL, AVIL,MIR25,GDF15,ADAMTS5,ABCA1,TACR1,PRICKLE1,NRP2,SIX4, FZD7,NTSR1, SYT2, SEMA6A, SIX1,DACT3, GPM6B, LOX,ISM1, GRIN1, ROBO2, RSAD2, P2RY1, GATM, HK2,SPRY1, KCNJ2,CACNA1G, DTX4, NPY1R,APLN, MCC, PTPRD, GDPD2, TNFRSF1B, DRAXIN, SYT7 |
Signaling |
11 |
GRIN1, KCNIP4, NODAL, AKAP5, NTSR1, PTPRD, ASIP,KCNJ2, GDF15, CACNA1G, SLC1A3 |
Feeding Behavior |
6 |
GRIN1, P2RY1, NPY1R, APLN,ASIP, GDF15 |
Regulation of synapse assembly |
5 |
ROBO2, GRIN1, SIX4, PTPRD,SIX1 |
Negative regulation of fibroblast growth factor receptor signaling pathway |
3 |
SHISA2, APLN,SPRY1 |
Cranial ganglion development |
3 |
NRP2, SIX4, SIX1 |
Trigeminal ganglion development |
3 |
NRP2, SIX4, SIX1 |
Regulation of animal organ formation |
3 |
ROBO2, SPRY1, SIX1 |
Table 2: Go term analysis with 190 top downregulated genes. A total of 190 downregulated genes with log2fold change of at least 1.5 were selected and analyzed using GOrilla (only 100 genes generated GO terms used in this table).

Figure 6: Schematic model of the proposed physiological changes with Pt(DECO)2 treatment in HeLa cells. All genes in yellow rectangles in the models are either upregulated or downregulated according to our RNAseq analysis. A) Pt(DECO)2 appears to upregulate genes responsible for cornification, nucleosome assembly, DNA damage response, p53 modulation, cytochrome activities in the ER, metabolism in the mitochondria, cell cycle control, and apoptosis. From all these processes, we selectively described the following cellular processes in the Discussion section: DNA damage response, p53 modulation, cell cycle control, and apoptosis. B) Several genes appear to be downregulated by the presence of Pt(DECO)2, including genes involved in drug transport, glutamate binding, transcription regulation, cell cycle progression, electron transport chain, mitochondria metabolism, anti-apoptosis, super elongation complex, DNA repair, and cell proliferation. We chose to provide more in-depth explanation on the following cellular processes in the discussion section: drug transport, cell cycle progression, transcription regulation, proliferation, super elongation complex and anti-apoptosis.
Chemotherapy remains one of the major therapeutic approaches treating a wide variety of cancer malignancies [20]. However, development of resistance to chemotherapy treatments and underlying broad spectrum of side effects of modern anti-cancer drugs have warranted a search for novel agents that could effectively work against cancer. The pursuit for finding an alternative metal based anti-cancer agent with limited side effects led to the synthesis and characterization of metal cyanoximates [8,9]. Only very few published studies value the prospects of newly prepared cyanoximates, specifically Pt(DECO)2 and Pd(DECO)2 [6,8,9]. Therefore, to our knowledge, we are one of the first to investigate their potentials as novel anti-cancer agents. Our present study is aimed at exploring the effects of our freshly synthesized metal cyanoximates on HeLa cancer cells. Our results herein provide valuable data and lay the groundwork for showing the promising effect of metal cyanoximates in cancer chemotherapy.
Effects of Metal cyanoximates on cell viabilityOne of the approaches that can be exploited for testing the potency of chemical compounds on cancer cells is the use of cell viability and proliferation assays [21]. For this study, we employed an XTT tetrazolium dye-based kit that offers a rapid and precise procedure to estimate cell proliferation and cytotoxicity [21,22]. Although the trypan blue assay has been described as a low-cost protocol, the method is associated with a high variance and lack of precision with results [23]. MTT assay uses tetrazolium salt known as 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) to measure cell proliferation and cytotoxicity [24]. The time-consuming washing step of solubilizing the MTT formazan crystal product in an organic solvent [25] makes the XTT assay more efficient to use. The present study revealed that Pt(DECO)2 reduces cell viability, comparable to the decreased level of cell viability caused by Cisplatin as shown by our XTT data. Additionally, our findings revealed that Pt(DECO)2 exhibits stronger anti-cancer functions against HeLa than Pd(DECO)2 and previously tested Pt(MCO)2. Pt(DECO)2 at 0.25mM concentration caused a 13% net reduction in cell viability, whereas Pd(DECO)2 and Pt(MCO)2 did not show lessened viability at the same concentration. Our finding is consistent with a previous investigation [6] that reported an excellent efficacy of platinum cyanoximates in regards to their activity against cancer cells. The study showed that treatment of 0.1 mM Pt(MCO)2 diminished HeLa and WiDr cell populations by 17% and 20%. However, the same concentration of Pd(DECO)2 reduced the cell population by only 5% and 15%, respectively. We attribute the better anti-proliferative effect of Pt(DECO)2 to the structural flexibility of the chemical, and this flexibility supports its thermodynamically stable state as well as its suitable kinetic disposition that mediates its faster solubility and better anti-cancer effect. The lesser biological activity of other cyanoximates can be linked to the increased bulkiness and rigidity of their anion, and therefore, there is higher energy barrier between the atom structure making it less flexible and more conformational (Personal communication with Dr Gerasimchuk from Missouri State University).
Effects of metal cyanoximates on reactive oxygen speciesOne of the mechanisms of action exhibited by chemotherapeutic agents is the generation of ROS to induce cell death [10]. In the current study, we observed that 0.1mM Cisplatin increased superoxide levels, which is in good agreement with previous findings that Cisplatin treatment elevates ROS concenrtation to levels above the threshold that causes cancer cell death [10,26-28]. On the contrary, we observed that under the same conditions, Cisplatin decreased the amount of peroxynitrite in treated HeLa cells. Taking into consideration the fact that the combination of nitric oxide with superoxide anion leads to formation of peroxynitrites [26,29], we conjecture that the decline in peroxynitrites could be due to a reduction in endogenous nitric oxide levels, but not superoxide due to our observation that superoxide levels increased with cisplatin (Figure 3). This idea is in line with the previous findings that have explained that reduction in the levels of endogenous nitric oxide in melanoma cell lines treated with Cisplatin correlated to depletion in cell growth and enhanced cisplatin induced apoptosis [30]. Likewise, a reduction in nitric oxide levels after Cisplatin treatment was linked to improved sensitivity and better response to Cisplatin, while elevated nitric oxide levels promoted cisplatin resistance [31,32]. As opposed to Cisplatin, our newly tested metal cyanoximates did not reveal significant alterations in both nitric oxide and superoxide levels, and we therefore, conclude that this may be due to a milder effect of the novel compounds compared to Cisplatin at the same conditions. Moreover, several kinds of ROS exist [33], and we are of the opinion that metal-cyanoximtaes may have a more noticeable impact on other types of ROS. At the same time, we cannot exclude the possibility that no alteration of ROS levels with Pt (DECO)2 could be due to a slight difference in mode of action between Cisplatin and tested metal cyanoximates.
Metal cyanoximates and apoptosis in HeLa cellsCisplatin, Carboplatin and Oxaliplatin are all platinum complexes currently used in multiple cancer treatments [27,34,35]. Our studies showed that all metal cyanoximates induce apoptosis in treated HeLa cells at 0.1 mM concentration. We postulate that tested Pt(DECO)2, Pd(DECO)2 and Pt(MCO)2 promoted apoptosis using a mechanism similar to currently used chemotherapeutics. The mostly commonly supported mechanism suggested by a growing body of evidence is that most anti-cancer drugs mediate apoptosis through the intrinsic mitochondria-dependent apoptotic pathway [36]. This pathway is believed to be initiated in response to intracellular stress signals such as DNA damage and excess ROS and is observed in most chemotherapy treated cells [37]. Studies have also revealed that metal based chemotherapeutic agents form covalent bonds with DNA, halting cell division and consequently inducing apoptosis [27,35]In order to understand the precise mode of action for the newly studied metal cyanoximates, we present a model below that contains highly upregulated and downregulated processes induced after Pt(DECO)2 treatment.
Upregulated gene expressions mediated by Pt(DECO)2 treatment and their proposed mechanism of actionHere, we provide a model (Figure 6A AND B) depicting the key upregulated genes and their corresponding cellular functions. From the model illustrated in Figure 6A, we highlighted the major processes upregulated after Pt(DECO)2 treatment. These processes include DNA damage response, p53 activation, cell cycle check point, and apoptosis (Figure 6A). PARP3 is known to be activated as a result of DNA damage response [38]. With the upregulation of PARP3 in our RNAseq analysis (Figure 6A), one can propose that Pt(DECO)2 treatment may cause significant DNA damage in HeLa cells. It has been known that the modulation of p53 activity plays a key role in DNA damage response [39]. Interestingly, our RNAseq analysis revealed that DAXX gene (death domain-associated protein), a well-known p53 regulator [40], was significantly upregulated. Further, a study has shown that DAXX binds to MDM2 (mouse double minute2 homolog), concurrently with Hausp (herpesvirus-associated ubiquitin-specific protease; also known as USP7) to stabilize the activity of MDM2 to modulate p53 function [41,42]. When cells are damaged or exposed to DNA damaging agents, the MDM2-DAXX interaction is distorted in an ATM-dependent manner, resulting in p53 activation [42,43]. Though the DNA damage induced by the treatment of Pt(DECO)2 in our experiment upregulates DAXX expression, the elevated DAXX may not secure MDM2 activity in this scenario, rather leading to hyper activation of p53 for cell death.
DNA damage is one of the prerequisite factors that promote cell cycle checkpoint system to slow down or halt the progression of cell division [44]. As cells enter a quiescent stage (G0), levels of (Growth arrest specific 7) GAS7 expression are elevated [45]. We found that Pt(DECO)2 treatment led to increased GAS7 expression (Figure 6A), suggesting that the treated cells are less likely proliferative. This was indeed the case proven by our XTT assay results (Figure 1). The level of GAS7 is also implicated in regulating the severity of metastasis, studies have demonstrated that lower level of GAS7 expression promotes tumor metastasis in several cancer types [46-48]. However, increased expression of GAS7 gene has been observed in Oxaliplatin-treated human hepatocellular carcinoma cells where it mediated proliferation reduction [47]. From these findings, we can infer that Pt(DECO)2-mediated GAS7 upregulation leads to decreased cell proliferation.
Elevated activity of p53 is associated with cell cycle arrest and apoptosis [49]. In agreement with this, RNAseq analysis revealed two highly expressed pro-apoptotic genes, CASP4 (caspase 4) and AIFM2 (Apoptosis-inducing factor, mitochondrion-associated 2) (Figure 6A). The former is shown to be upregulated in response to endoplasmic reticulum stress induced via an increased number of unfolded and misfolded proteins [50-52]. Additionally, a recent study showed that decreased CASP4 expression is associated with poor prognosis of esophageal squamous cell carcinoma. This finding supports that notion that CASP4 promotes inflammatory and immune response that may inhibit squamous cell carcinoma progression [52]. AIFM2 has been associated with mitochondria-mediated apoptosis signaling [53], and its upregulation promotes apoptosis in lung cancer cells [54]. CASP4 or AIFM2 can serve as an upstream apoptotic mediator to enhance CASP9 activation [55,56], and Caspase 9 activation subsequently initiates caspase‐3 activation, which eventually activates the rest of the caspase cascade and promotes apoptosis [57]. Taken together, we propose that Pt(DECO)2 treatment stimulates apoptosis in a caspase dependent manner.
Downregulated Genes mediated by Pt(DECO)2 treatment and their proposed mechanism of actionBased on the GO terms list generated from GOrilla(Figure 6B), mulitple highly downregulated genes were identified in response to Pt(DECO)2 treatment. The most downregulated genes were responsible for drug transport, cell cycle progression, transcription regulation, proliferation, super elongation complex and anti-apoptosis (Figure 6B). SLC47A2, SLC1A3, SLC16A9, ABCA1, ABCA3, and ABCA12 are all classified as drug transporters and are downregulated in our model (Figure 6B). Drug transporters in cells are classified into solute carriers (SLC), and ATP-binding cassettes (ABC) transporters [58,59]. Evidence has shown that cancer cells utilizes the effect of SLC to promote growth and survival [58,60]. ABC transporters are efflux drug transporters that utilizes energy from ATP hydrolysis to pump toxic drugs out against their concentration gradient. Increased expression of several ABC transporter genes is associated with reduced cellular accumulation of anticancer agents and acquired drug resistance in treated cell lines [58,59]. Evidence has shown that cancer cells utilize the effect of SLC to promote growth and survival [58,60]. ABC transporters are efflux drug transporters that utilize energy from ATP hydrolysis to pump toxic drugs out against their concentration gradient. Increased expression of several ABC transporter genes are associated with reduced cellular accumulation of anticancer agents and acquired drug resistance in treated cell lines [58,59]. Thus, Pt(DECO)2 treatment enhances downregulation of drug transporter genes that may otherwise induce drug resistance in treated cancer cells.
SIX1 and SIX4 are members of Sine Oculis Homeobox (SIX) homolog family that has historically been implicated in regulating transcription. Their upregulation is thought to result in tumor metastasis, growth, and invasion in non-small cell lung cancer [61]. In contrast, our result of downregulation of SIX1 and SIX4 by Pt(DECO)2 [62], is suggestive of a role in decreasing cancer proliferation.
NODAL gene is a member of the TGF-β family of proteins that is essential for development of embryonic cells [63,64]. Recently, findings have elucidated that NODAL gene is highly expressed in multiple cancer types and functions by promoting carcinogenesis [63,64]. Our model displayed that Pt(DECO)2 treatment reduced the expression of NODAL gene as illustrated in Figure 6B. Downregulation of the NODAL gene provides evidence that Pt(DECO)2 fosters the reduction of viability in cancer cells by suppressing the genes that enhances rapid growth and proliferation.
Furthermore, BCL2, a well-known anti-apoptotic gene commonly overexpressed in several cancer types was also found to be downregulated [65]. Notably, the anti-apoptotic potential of BCL2 has also been correlated to chemotherapy resistance, with findings suggesting that overexpression of this genes makes it rather difficult to eliminate malignant cells with cytotoxic anticancer agents [66]. Downregulation of BCL2 genes by Pt(DECO)2 further supports its role in promoting apoptosis. We also noticed the downregulation of a key component of the super elongation complex (SEC), AFF4 gene (Figure 6B). The elongation stage of transcription is regulated by a complex group of genes that comprise the super elongation complex (SEC). The product of the AFF4 gene directly binds with P-TEFb and AF9 or ENL and is needed for the formation of SEC [67,68]. Overexpression of the AFF4 gene has been shown to promote cell growth and proliferation in head and neck cell squamous cell carcinoma (HNSCC) [69]. Our model revealed that AFF4 was downregulated, implying that Pt(DECO)2 can help reduce cancer cell proliferation.
Genes that maintain the cell cycle have been shown to promote rapid growth and proliferation in cancer cells [70]. Our model indicates that GTSE1, ESCO2, NPAT, and E2F8 genes were downregulated (Figure 6B). E2F8 is a key member of the E2F family of transcription factors that have demonstrated key roles in cell growth, differentiation and cycle regulation [71]. E2F8 over expression has been noted in hepatocellular carcinoma (HCC) affirming its role in tumorigenesis [72]. Likewise, GTSE1 is upregulated in several human cancers owing to the fact that it negatively regulates p53 expression [73,74]. A recent study, however, established that GTSE1 overexpression in hepatocellular carcinoma (HCC) promoted the metastasis and proliferation in the cell line [73]. Both GTSE1 and E2F8 were downregulated in our model, declining their functions in HeLa cells treated with Pt(DECO)2. We deduced from our model that Pt(DECO)2 suppresses activities of drug transporters, anti-apoptotic genes, genes that promotes proliferation as well as genes that represses apoptosis. The overall findings highlight the roles of Pt(DECO)2 as a potential anti-cancer agent through different activities of differentially expressed genes and their several roles in reducing cell proliferation and promoting apoptosis.
In the present study, we assessed the anti-cancer potentials of newly synthesized metal cyanoximates on HeLa cells and provided evidence that treatment with metal cyanoximates enhanced reduced viability and apoptosis. Furthermore, we selected Pt(DECO)2 as the most effective in the group and performed RNA sequence analysis to establish its mode of action. Differential expression of several hundred genes that play key roles in cell division, cell cycle control, proliferation and apoptosis supported our initial studies that Pt(DECO)2 augments cell death. These novel findings lead us to conclude that Pt(DECO)2 can serve as a potential alternative for use in cancer chemotherapy.
We are indebted to Dr Nikolay Gerasimchuk in the chemistry department at Missouri State University for productive collaborations and generously providing prepared metal cyanoximates used for this study. We also appreciate the graduate thesis funding provided by the Graduate College and the grant for RNA sequence analysis that was provided by the College of Natural and Applied Sciences (CNAS) at Missouri State University.
The authors declare no conflict of interest.
Citation: Kafayat Aderonke Yusuf and Kyoungtae Kim. “Novel Cyanoximates as Chemotherapeutic Candidates". Acta Scientific Microbiology 4.4 (2021): 119-134.
Copyright: © 2021 Kafayat Aderonke Yusuf and Kyoungtae Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

















 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
