Nyasha Chin’ombe1*, Boniface Muzividzi2 and Pasipanodya Nziramasanga1
1Molecular Microbiology Laboratory, Department of Medical Microbiology, University of Zimbabwe, Avondale, Harare, Zimbabwe
1National Microbiology Reference Laboratory, Ministry of Health and Childcare, Southerton, Harare, Zimbabwe
*Corresponding Author: Nyasha Chin’ombe, Molecular Microbiology Laboratory, Department of Medical Microbiology, University of Zimbabwe, Avondale, Harare, Zimbabwe.
Received: September 09, 2020; Published: March 22, 2021
Background: Nontuberculous Mycobacteria (NTM) are now commonly detected in humans and are ubiquitously distributed in various environments such as water, soil and air. Limited data exist on the diversity and accurate identification of NTM species in Zimbabwe. This study evaluated the use of 16S-23S rRNA ITS in genetically characterizing the diversity of NTM isolated from Zimbabwe during a national tuberculosis survey.
Methods: Polymerase chain reaction was used to amplify the 16S-23S rRNA ITS DNA from archived NTM isolates from National Microbiology Reference Laboratory (NMRL) of the Ministry of Health and Childcare, Zimbabwe. The amplicons were sequenced and analysed using bioinformatics tools.
Results: From the 963 archived NTM isolates at the NMRL, 26 had their 16S-23S rRNA ITS DNA sequences analysis. Genetic heterogeneity of the 16S-23S rRNA ITS was demonstrated among the NTM isolates from Zimbabwe. The analysis showed that there were 15 (57.7%) M. avium, 4 (15.4%) M. palustre, 2 (7.7%) M. seoulense, 2 (7.7%) M. parascrofulaceum, 1 (3.8%) M. sinense, 1 (3.8%) M. asiaticum and 1 (3.8%) M. bouchedurhonense.
Conclusion: The study demonstrated that the 16S-23S ITS region could be used in characterizing NTM species from Zimbabwe. Most of the NTM isolates from Zimbabwe were closely related to M. avium, a known human opportunistic pathogen. Further studies are however necessary to unravel the true scope of NTM problem in Zimbabwe.
Keywords: Nontuberculous Mycobacteria; 16S-23S rRNA ITS; Diversity; Zimbabwe
Nontuberculous Mycobacteria (NTM) represent a very diverse group of microorganisms that are ubiquitously distributed in several environments such as water, soil, sewage and even hospital settings [1]. More than 170 of species of NTM have so far been identified and most of them are known to cause opportunistic and non-opportunistic infections in humans [2,3]. Identification and differentiation of the different species of NTMs is critical for successful treatment and management of infections. Traditional methods such as biochemical tests and chromatographic method are not reliable for identification of these microorganisms. In the past few years, molecular methods have been used in identification and differentiation of NTM [4]. These methods allow the easy differentiation and identification of Mycobacterial species. Amplification and sequencing of the 16S ribosomal RNA gene (rDNA) is regarded as the golden standard method for identification of NTM [5]. Other genes are also available for use in NTM identification by amplification and sequence analysis. The intergenic spacer region that lies between the 16S and 23S ribosomal RNA gene is hyper-variable in most bacterial species and can potentially be useful in NTM characterization and speciation [6]. In Zimbabwe, there is scarce data on genetic characteristics and diversity of NTM and there is also lack of proper and reliable NTM identification methods in clinical and non-clinical settings.
The aim of the study was therefore to investigate the use of 16S-23S rRNA ITS in genetic characterization and identification of NTM isolates from Zimbabwe.
The samples used in this study were archived NTM isolates at the National Microbiology Reference Laboratory of the Ministry of Health and Childcare (Government of Zimbabwe). The history of the archived NTM samples was previously reported [7]. The bacterial cultures were retrieved and recultured and the DNA was extracted as previously described [7].
Amplification of the 16S-23S rRNA ITS DNAThe 16S-23S ITS DNA of the NTM isolates was amplified by polymerase chain reaction using the GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA). The Mycobacterium-specific forward primer (16SC), 5'- TCG AAG GTG GGA TCG GC -3' and reverse primer (23SC), 5'- GCG CCC TTA GAC ACT TAC-3' were used and an approximately 380 bp PCR product was expected to be amplified [8]. The polymerase chain reaction contained 36.8 μl of distilled water, 0.2 μl of Dream Taq polymerase (5 U/μl stock) (Inqaba, South Africa), 1 μl of dNTPs (10 mM stock) (Inqaba, South Africa), 1 μl forward primer (10 μM stock), 1 μl reverse primer (10 μM stock), 5 μl of 10x PCR Dream Buffer and 5 μl of template DNA to make a total volume of 50 μl. The polymerase chain reaction was performed using the following cycling profile: initial denaturation for 5 minutes at 950C, followed by 45 amplification cycles of 30 seconds at 950C, 30 seconds at 540C, 30 seconds at 720C and ending with one cycle of final extension step for 5 minutes at 720C. The amplification (10 μl of PCR products) was checked by 2.5% agarose gel electrophoresis.
Amplicon DNA sequencingThe DNA amplicons that showed gel bands of the expected size were selected for sequencing. They were sent to Inqaba Biotechnical Industries, Pretoria (South Africa) for sequencing according to standard sequencing protocols. Raw sequence data were emailed back to the researchers and analyzed using bioinformatics tools.
DNA sequence analysisAnalysis of the 16S-23S ITS DNA sequences was performed using common bioinformatics softwares, mainly Geneious Basic program (Biomatters, USA) and Basic Local Alignment Search Tool (BLAST) programs. The sequences were aligned to each other and were also compared to those in the Genbank database. Phylogenetic analysis of the DNA sequences was also performed and phylogenetic tree drawn using the Geneious Basic program. Attempts were made to identify the NTM isolates to species level using the sequence analysis. The sequences were aligned and compared with those in the Genbank using Geneious Basic program.
Details of the characteristics of the NTM samples used in this study were previously reported [7]. Briefly, this was a the cross-sectional study in which we took DNA extracted from a 99 NTM samples which were part of the 963 NTM samples isolated throughout Zimbabwe during the national tuberculosis prevalence survey in 2014 [7]. In this study, we amplified the 16S-23S ITS DNA from 48 of the 99 samples.
Amplicons of the 16S-23S intergenic spacer DNAOut of the 48 reactions, amplification was successful in 43 samples. On gel electrophoresis, the bands were strong in 42 of the 43 samples. Figure 1 shows a representative gel of the amplification of the intergenic region of the NTM samples. Some bands were larger than others, illustrating variation in the size of the intergenic region in different species of NTM. The sizes of the bands were different in sizes for some NTM isolates (Figure 1). The expected DNA band was approximately 380 bp, but this could range from 332 to 534 bp in size [9].
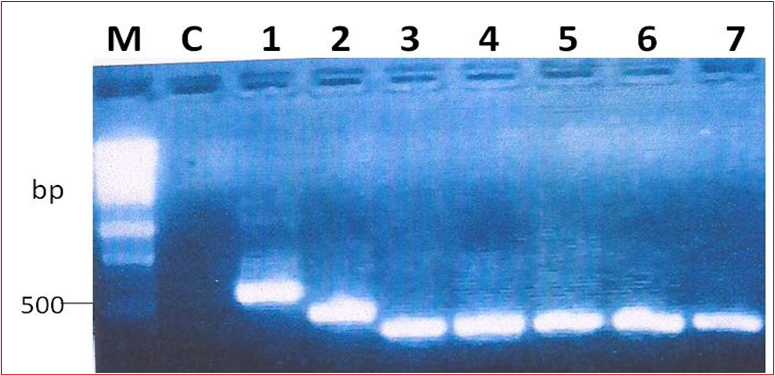
Figure 1: A representative gel of the amplification of the 16S-23S ITS DNA from NTM isolates. M is the molecular weight marker (Promega™, Wisconsin); Lane NC; negative PCR control, lanes 1-7 show positive 16S-23S ITSDNA amplicons.
A total of 43 amplicons sent for sequencing, only 26 were successfully sequenced and analysed using bioinformatics tools. The sequences were analysed for genetic diversity. Out of these, 23 of them were genetically unique and diverse (Figure 2). When the sequences were aligned to each other, it was shown that they differed even in length. Some were longer than others (Figure 2). Several nucleotide polymorphisms between the different isolates were also noted (Figure 2). Overall, genetic heterogeneity of the 16S-23S rRNA ITS existed among our NTM isolates from Zimbabwe. The sequencing also showed that spacer region of most of the NTM isolates differed in size.
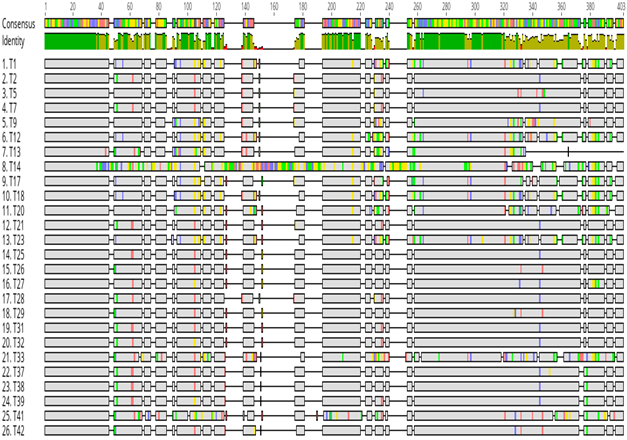
Figure 2: Genetic diversity of NTM isolates from Zimbabwe based on the 16S-23S ITS DNA. All the NTM sequences were aligned and the conserved (in grey colour) and the hypervariable regions of the 16S-23S ITS DNA were shown. The grey areas represent the conserved regions, the hypervariable regions are represented by blue (Cytosine), green (Adenine), red (Thiamine) and black (Guanine) regions in the 16S-23S ITS DNA.
Further analysis of the 16S-23S ITS DNA sequences was performed in order to identify them to species level. The sequences were aligned to each other and those from the Genbank database. The analysis of the 26 showed that all the isolates belonged to the Genus Mycobacterium (Table 1). Out of the 26 NTM isolates, there were 15 (57.7%) M. avium, 4 (15.4%) M. palustre, 2 (7.7%) M. seoulense, 2 (7.7%) M. parascrofulaceum, 1 (3.8%) M. sinense, 1 (3.8%) M. asiaticum and 1 (3.8%) M. bouchedurhonense (Table 2). A phylogenetic tree was constructed to show the close relationship of all the NTM isolates from Zimbabwe and their close relates from the Genbank database (Figure 3). M. tuberculosis and M. intracellulare sequences were included as controls.
| Sample Sequence ID Number | Mycobacterium Species Identification based on ITS % similarity | % similarity |
|---|---|---|
T1 |
M. palustre (HM454240) |
99.5 |
T2 |
M. avium (FJ858758) |
100 |
T5 |
M. avium (FJ858759) |
98.4 |
T7 |
M. avium (FJ858758) |
100 |
T9 |
M. parascrofulaceum (FJ858765) |
99.2 |
T12 |
M. palustre (HM454240) |
98.2 |
T13 |
M. seoulense (L15622) |
95.8 |
T14 |
M. sinense (CP002329) |
94.8 |
T17 |
M. parascrofulaceum (AY337282) |
96,2 |
T18 |
M. palustre (HM454240) |
99.5 |
T20 |
M. seoulense (FJ042511) |
95.1 |
T21 |
M. avium (FJ858758) |
99.7 |
T23 |
M. palustre (HM454240) |
98,7 |
T25 |
M. avium (EU266629) |
99.5 |
T26 |
M. avium (FJ858759) |
98.7 |
T27 |
M. bouchedurhonense (EF591051) |
99.0 |
T28 |
M. avium (FJ858758) |
100 |
T29 |
M. avium (FJ858759) |
98.4 |
T31 |
M. avium (FJ858759) |
99.8 |
T32 |
M. avium (FJ858758) |
99.8 |
T33 |
M. asiaticum (MF774081) |
99.4 |
T37 |
M. avium (EU266629) |
99.5 |
T38 |
M. avium (EU266629) |
99.7 |
T39 |
M. avium (FJ858758) |
99.7 |
T41 |
M. avium (FJ858758) |
92.9 |
T42 |
M. avium (FJ858759) |
98.6 |
Table 1: Identification of NTM species based on 16S-23S ITS sequence analysis (n = 26).
| NTM species | No. | Frequency (%) |
|---|---|---|
M. avium M. palustre M. seoulense M. parascrofulaceum M. sinense M. asiaticum M. bouchedurhonense |
15 4 2 2 1 1 1 |
57.7 15.4 7.7 7.7 3.8 3.8 3.8 |
Table 2: NTM species identified based on 16S-23S ITS sequence analysis (n = 26).
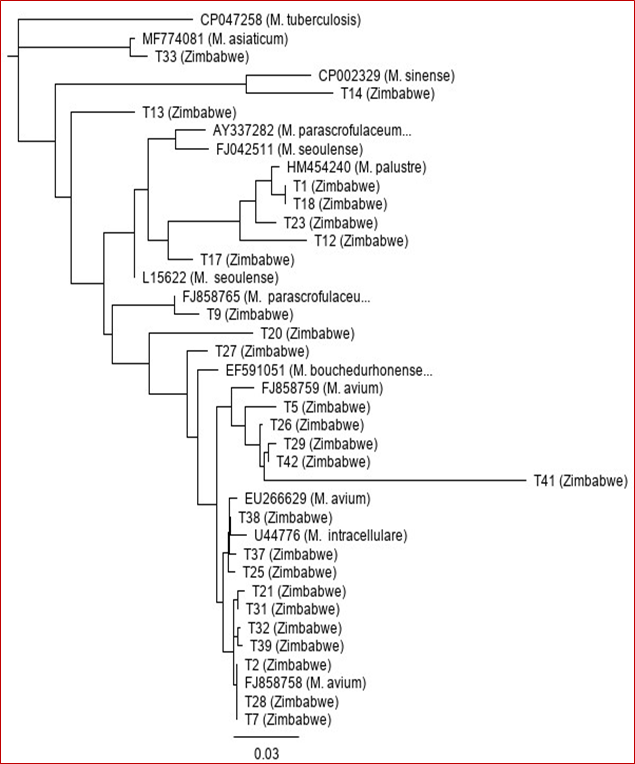
Figure 3: Neighbour-joining phylogenetic tree based on the 16S-23S ITS DNA sequences showing genetic relationships of NTM isolates from Zimbabwe and the closely related species from the Genbank database. The tree was rooted with M. tuberculosis 16S-23S ITS DNA sequence (Accession number CP047258).
The prevalence of nontuberculous Mycobacteria has increased globally in the past few years [10]. These diverse bacteria are now being detected frequently in clinical specimens as well as in various environments in several countries [11,12]. They cause a spectrum of diseases such as pulmonary and extrapulmonary disease, cervical lymphadenitis in young children, and visceral and disseminated disease [13]. Accurate and reliable identification of the NTM is therefore key. Traditional approaches and strategies are time-consuming and labour intensive and are unreliable [14]. Molecular methods which are more reliable and accurate have been developed for the accurate identification of the different NTM species [15]. Targeting and sequencing specific genes is now one of the strategies. The 16S ribosomal RNA gene is the mostly used gene because of its genetic conservation within different NTM species and diversity among species [5]. The intergenic region that lies between 16S and the 23S ribosomal RNA genes is known to be highly variable in bacteria at species or sub-species level and has therefore been proposed for use in genetic analysis of various taxonomic groups [16]. In this study, we investigated the sequence diversity of the 16S-23S intergenic spacer DNA region with the possibility of using the data in identification of NTM isolates from Zimbabwe. The amplification of the 16S-23S intergenic spacer DNA using polymerase chain reaction showed that some species had different sizes of this region (Figure 1). The 16S-23S ITS region in NTM differs in size and its genetic variation from species to species makes it suitable for NTM differentiation [17]. Some isolates had shorter intergenic region and others had slightly longer region. This result was expected. It is generally known that the size of the intergenic spacer DNA which lies between the 16S and 23S ribosomal RNA genes [9]. Depending on bacterial species, the length of the 16S-23S rRNA ITS regions where normally the tRNA-encoding genes are found varies considerably from 200 - 1500 bp [16]. This variation and diversity has resulted in bacterial taxonomist to use 16S-23S rRNA ITS in differentiating species even to strain level [18]. Several studies have demonstrated the utility of 16S-23S rRNA ITS in differentiation of NTM even those which are closely related [9,19-23]. We were also able to find species from the Genbank database that were closely related to our NTM isolates from Zimbabwe (Figure 3). A phylogenetic analysis showed that our isolates were closely related to NTM that have been isolated elsewhere. We found M. avium, M. palustre, M. seoulense, M. parascrofulaceum, M. sinense, M. asiaticum and M. bouchedurhonense. The majority of the isolates belonged to M. avium and this was in line with findings from our previous study in which we used 16S rRNA gene-based method and found that the majority of the NTM isolates belonged to M. avium complex species [7]. M. avium is known to cause avian tuberculosis in almost all avian species and is now one of the most common opportunistic pathogen in humans [24]. It is ubiquitously distributed in various environments. M. palustre is a slow-growing human pathogen that has been isolated in several clinical settings [25]. M. seoulense and M. parascrofulaceum were previously isolated from patients with pulmonary infections [26,27]. M. sinense was recently isolated from a patient with severe lung infiltrates and the bacterium was found to be resistant to most Mtb drugs [28]. M. asiaticum is associated with pulmonary disease, lymphadenitis in children and wound infections [29]. M. bouchedurhonense was recently described and is regarded as a new species in the M. avium complex group [30].
The study demonstrated that the 16S-23S ITS region could be used in characterizing NTM species from Zimbabwe. Further studies are however necessary to unravel the true scope of NTM problem in Zimbabwe
Conflict of InterestThe authors confirm that this article content has no conflict of interest.
AcknowledgementsThe authors thank staff from the Departments of the Medical Microbiology and Biological Sciences of the University of Zimbabwe and the National Microbiology Reference Laboratory for support during the study. The research study was supported by a grant from the University of Zimbabwe’s Research Board (Grant Code 91065).
Citation: Nyasha Chin’ombe., et al. “16S-23S Ribosomal RNA Internal Transcribed Spacer Region-based Genetic Analysis of Nontuberculous Mycobacteriafrom Zimbabwe". Acta Scientific Microbiology 4.4 (2021): 64-69.
Copyright: © 2021 Nyasha Chin’ombe., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

















 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
