Ilias Jerrar Oulidi1, Fatima Zahrae Benchekroun2*, Wafae Chouhani3 and Chakib Nihal4
1Department of Internal Medicine, MUSC Health, USA
2Faculty of Medicine, Dentistry and Pharmacy of Fez, University of Sidi Mohamed
Ben Abdellah, Morocco
3Department of Neurology, Trinity Health Grand Rapids, USA
4Centre Al Hamd De Dialyse Et De Maladies Rénales, Morocco
*Corresponding Author: Fatima Zahrae Benchekroun, Faculty of Medicine, Dentistry and Pharmacy of Fez, University of Sidi Mohamed Ben Abdellah, Morocco.
Received: August 12, 2024; Published: September 06, 2024
Citation: Fatima Zahrae Benchekroun., et al. “Acute Kidney Injury Revealing Adult-Onset Still’s Disease". Acta Scientific Clinical Case Reports 8.10 (2024):13-17.
Adult-onset Still's disease (AOSD) is a rare systemic inflammatory disease of unknown etiology. That has been documented in literature with respect to its associated complications, including but not limited to cardiac, pulmonary, hematological, hepatic, and albeit rarely, renal complications.
We present the case of a woman in her mid-sixties who presented with fever, generalized rash, pharyngitis, and inflammatory polyarthralgia. Further laboratory investigations indicated the presence of acute renal failure (ARF) of the acute tubulointerstitial nephritis type (ATIN), ultimately leading to an AOSD diagnosis. The patient was promptly administered oral corticosteroids, resulting in a positive clinical and biological outcome. Attributing renal damage to AOSD was retained after all other potential causes of ATIN were ruled out. The diagnosis was further confirmed by observing a favorable response to treatment.
Keywords: Adult-onset Still's Disease (AOSD); Acute Renal Failure (ARF); Acute Tubulointerstitial Nephritis (ATIN); Systemic Inflammatory Disease; Corticosteroid Treatment
ALP: Alkaline Phosphatase; ANA: Antinuclear Antibodies; ANCA: Antineutrophil Cytoplasmic Antibodies; Anti-CCP: Anti-Cyclic Citrullinated Peptide; AOSD: Adult-onset Still's Disease; ATIN: Acute Tubulointerstitial Nephritis; AKI: Acute Kidney Injury; BUN: Blood Urea Nitrogen; CRP: C-Reactive Protein; DIC: Disseminated Intravascular Coagulation; ESRD: End-Stage Renal Disease; GGT: Gamma-Glutamyl Transferase; Hb: Hemoglobin; MGL: Minimal Change Glomerulopathy; MPA: Microscopic Polyangiitis; RHL: Reactive Hemophagocytic Lymphohistiocytosis; SPEP: Serum Protein Electrophoresis; TMA: Thrombotic Microangiopathy; WBC: White Blood Cells
AOSD is a systemic inflammatory disease of unknown etiology that was first described by Bywaters in 1971 [1]. It is a rare condition with an incidence that varies from 0.16 to 0.4 cases per 100,000 inhabitants per year [2]. Diagnosis is based on the presence of Yamaguchi’s criteria [3] or Fautrel criteria [4] as well as the exclusion of other potential causes including infectious, inflammatory, and neoplastic etiologies. The clinical manifestations are characterized by a triad of hectic fever, inflammatory polyarthritis, and a maculopapular rash. Renal manifestations directly related to AOSD are exceptional, apart from isolated transient proteinuria during febrile episodes [5]. In AOSD renal damage, especially glomerular damage, can occur as a result of other complications such as amyloidosis AA, thrombotic microangiopathy (TMA), disseminated intravascular coagulation (DIC), or Reactive hemophagocytic lymphohistiocytosis (RHL) [5-7].
We describe the case of a patient who presented with acute renal failure, which ultimately led to the diagnosis of Adult-onset Still's Disease (AOSD).
A patient in her 60s, Gravida 8 Para 8, with a history of venous insufficiency on venotonics, presented with a ten-day history of fever around 38.5-39 °C, pharyngitis, non-pruritic generalized maculopapular rash [Figure 1,2], and inflammatory-like polyarthralgia.
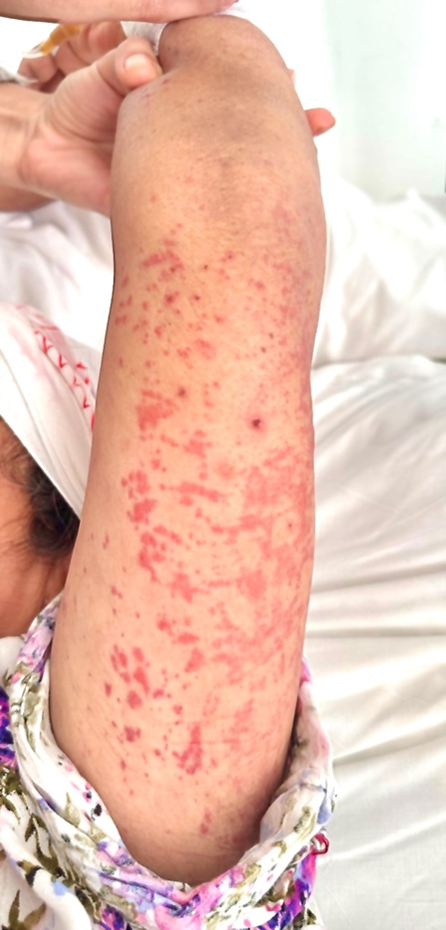
Figure 1: Image representing our patient’s upper limb maculopapular rash.

Figure 2: Image representing our patient’s inferior extremities rash.
She was hospitalized for 8 days for work up. On examination we noted a non-pitting edema of the lower limbs extending to the knees, as well as pleural effusions at the lung bases. On urine dipstick: protein was 1+, leucocytes 1 + and there was no hematuria.
Our Initial Approach, was to underwent a diagnostic work-up to rule out infectious, toxic, medication-related, and neoplastic causes (Table 1). Starting with urine analysis, it revealed positive proteinuria, aseptic leukocyturia, no hematuria, indicative of tubulo-interstitial nephropathy syndrome. Additionally, we ordered an initial complete Blood count, basic metabolic panel and comprehensive metabolic panel that showed acute kidney injury (AKI) with a high BUN and creatinine levels, hypercalcemia, no anemia. The patient showed signs of biological inflammatory syndrome with hyperleukocytosis and neutrophilic predominance without hypereosinophilia, a high CRP, hypoalbuminemia, hyper-alpha 1 globulinemia, and hyperferritinemia. Additionally, the patient had hepatic cholestasis. Procalcitonin was normal and serum protein electrophoresis (SPEP) did not show gammapathy. The immunological assessment was negative for ANCA, anti-CCP, antinuclear antibodies, and soluble antinuclear antigens.

Table 1: Pre-Treatment Laboratory Results.
On ultrasound kidneys had a normal size and were well-differentiated. The patient also had with pleuro-pericarditis as evidenced by echocardiography and chest CT. In the context of febrile tubulo-interstitial nephritis, we initially excluded an infectious source, particularly pyelonephritis. Pathogens were not detected in the urine culture. Further evaluation for infectious causes encompassed blood cultures, Quantiferon testing, and hepatic serologies, all were negative. The clinical assessment failed to pinpoint any localized infectious focus. The patient's medical history did not reveal the use of nephrotoxic medications, exposure to plants, or administration of contrast agents. Given the systemic elements evident in the clinical presentation – encompassing arthralgias, fever, renal involvement, and pleuro-pericarditis – lupus emerged as a prominent primary differential diagnosis. This was ruled-out by an immunological panel including ANCA, anti-CCP, antinuclear antibodies, and soluble antinuclear antigens. Although neoplastic origins were considered due to the prolonged fever and age, a comprehensive clinical examination, coupled with normal chest, abdominal and pelvic CT scans, absence of monoclonal spikes in protein electrophoresis, and a generally well-preserved state of health, subsequently led to the exclusion of this possibility.
Our therapeutic approach for this patient was to prescribe oral corticosteroid therapy at a dose of 0.5mg/Kg/day. In the course of one and a half month and over the in light of positive clinical and biological improvements, negative proteinuria, leukocyturia, and CRP levels, and a gradual normalization of renal function, steroids were gradually tapered. At three months assessment post-treatment, the patient’s serum ferritin and liver cholestasis returned to normal. However, dyspnea and basithoracic crackles persisted during subsequent follow-up consultations. The patient underwent corticosteroid therapy for a total of 5 months. After one year, the patient’s progress was favorable, and both the transthoracic echocardiogram and chest-X-ray revealed no abnormalities.
Our patient was diagnosed with AOSD using Yamaguchi criteria [3]. She presented the following major criteria: fever, arthralgia, typical rash, hyperleukocytosis (more than 80% of neutrophils), and minor criteria: pharyngitis, abnormal liver function tests, negative ANA, after ruling out the exclusion criteria. The patient’s AOSD was complicated by pleuro-pericarditis and an ATIN resulting in AKI. AOSD was retained as the cause of the ATIN after the exclusion of other possible etiologies including infectious, toxic, medication side effects, immunological or neoplastic. Renal involvement in AOSD is uncommon and polymorphic [5]. Renal amyloidosis type AA is the most commonly described renal complication in the literature [8-11,13].
When it occurs during AOSD in the absence of infectious, inflammatory, or tumoral pathologies, it is likely linked to the disease in question [13]. Its pathogenesis remains hypothetical, possibly multifactorial including the chronic elevation of “Serum Amyloid Associated Protein”, associated with genetic and environmental factors that are still unknown [11,12]. Renal amyloidosis in AOSD usually causes glomerular damage, characterized by mild proteinuria or a nephrotic syndrome, which can progress to end-stage chronic renal disease [13,14]. Vascular nephropathy which would be linked to the obstruction of the vessels by amyloid deposits leading to progressive ischemia of the renal parenchyma and causing progressive renal failure without proteinuria [8]. Other types of glomerulopathies have been described during the course of AOSD. These include cases of membranoproliferative glomerulonephritis [15], pauci-immune extra-capillary proliferative glomerulonephritis strongly suggestive of MPA (microscopic polyangiitis) [16], minimal change disease [17], segmental and focal collapsing hyalinosis [18,19] glomerular nephropathy with mesangial deposits of IgA [20].
In our patient, kidney biopsy was not performed given the rapid improvement after corticosteroid administration. On the other hand, tubulointerstitial nephropathy during AOSD has not been described or only rarely in the literature. We cite the case reported by Samuel Blas Gómez., et al. [21] of an AOSD discovered following an AKI, the biopsy of which came back in favor of IgA nephropathy associated with tubulointerstitial damage.
Therapeutic management of renal impairment in AOSD is not standardized. It therefore resembles the overall management of AOSD which relies on corticosteroid therapy [22]. The reported case of Gomez., et al. was treated with corticosteroid therapy in combination with Anakinra resulting in a rapid and favorable outcome [21]. We mention other types of renal damage, all treated with corticosteroid therapy: MGL [17], segmental and focal collapsing hyalinosis [18], and chronic glomerulonephritis (in addition to methotrexate) [23]. When corticosteroid therapy does not lead to a favorable improvement, the authors have used several immunosuppressive protocols with a very variable prognosis from one study to another, most often unfavorable with the evolution towards ESRD with the need for dialysis or death of patients.
Renal involvement in AOSD is uncommon but significant, this is why it is important to consider AOSD in the differential diagnosis of acute renal failure with systemic symptoms. When early recognized, corticosteroid therapy is crucial for managing this rare complication.
Copyright: © 2024 Fatima Zahrae Benchekroun., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff







